一种用于抗体药物偶联物的连接子及其应用的制作方法
本发明涉及一种用于抗体药物偶联物的连接子以及由该连接子制备的抗体药物偶联物,以及该抗体药物偶联物在治疗肿瘤或其它疾病中的用途。
背景技术:
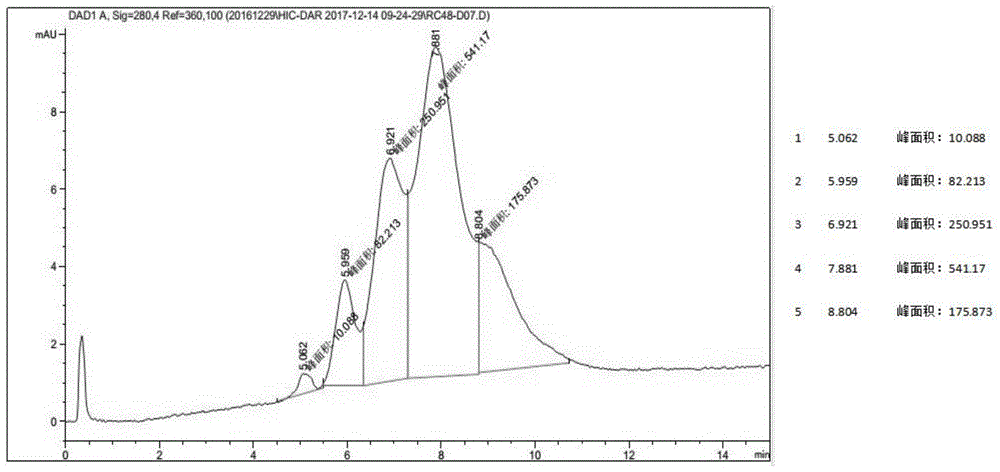
:抗体药物偶联物(antibody-drugconjugates,adcs)是一类靶向治疗药物,主要应用于癌症、自身免疫性疾病等治疗领域。其结构通常由三部分组成:抗体或抗体类配体、药物部分、以及将抗体或抗体类配体及药物偶联起来的连接子(linker)。传统的adc药物是将药物通过连接子偶联到抗体的赖氨酸残基或者是链间二硫键还原而产生的半胱氨酸残基上。当选用赖氨酸残基作为偶联位点时,由于一个抗体大约含有40个以上的赖氨酸残基,且偶联反应非选择性,偶联数目和位点具有极大的不确定性,产品均一性非常差;当选用半胱氨酸残基作为偶联位点时,尽管抗体(igg1)只有4对链间二硫键,通过还原链间二硫键产生的半胱氨酸残基形成的adc药物的dar(药物抗体偶联比率)值较均一,但是此种偶联方式破坏了抗体的二硫键,影响了抗体的稳定性,并且由于现有还原剂(dtt、tcep)对链间二硫键的选择性较差,因此最终产物均一性也较差。而由于治疗效果和药品监管的原因,产物的均一性很重要。因此,需要制备具有药物抗体偶联比率控制良好的adc药物。近年来,有研究者运用基因工程技术将抗体进行改造,从而实现抗体与药物的定点偶联,所得到的adc也具有十分均一的dar值,但是因为基因重组技术需要大量的工作和设计以寻找适宜的位点供药物偶联或聚乙二醇修饰,因此非常耗时而且研发费用极高。针对上述现有技术中制备抗体药物偶联物的缺陷,本领域迫切需要开发新的连接子,以实现毒性药物定点偶联抗体,从而获得产物均一性更高、稳定性更强的adc药物。技术实现要素:为了解决上述问题,本发明提供了一种用于制备抗体药物偶联物的连接子以及由该连接子制备的抗体药物偶联物,以及其在治疗肿瘤药物中的用途。所述的连接子可同时与抗体或者抗体功能性片段上的巯基或者氨基偶联,尤其是能够与抗体或者抗体功能性片段上的2、3或4个巯基偶联,偶联后的产物均一、结构稳定。在本发明的第一方面,提供了一种连接子,其结构为式i所示:其中,m1'、m2'各自独立的选自或为空,m1'、m2'相同或不同,但不同时为空;x、y、r1、l1、l2、l3'、q为任意的基团。进一步的,前述所述的x、y、r1、l1、l2、l3'、q可以为:l1、l2各自独立地选自c1-c9烷基、c2-c9烯基、c2-c9炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基、聚乙二醇、o、s、及其任意组合;q选自n、b、p、芳基、杂芳基、环烷基、(杂环基)烷基;l3'选自聚乙二醇、及其任意组合;x选自f、cl、br、i;y为(上述两个结构中,r2可在环上的任意位置取代。)z选自c、n;r1、r2、r3、r4、r5、r8、r10、r11、r12、r13、r14、r15各自独立地选自h、c1-c6烷基、c2-c6烯基、c2-c6炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基。更进一步的,前述所述的x、y、r1、l1、l2、l3'、q可以为:l1、l2各自独立地选自c1-c4烷基、c6-c8芳基、c6-c8杂芳基、c5-c6环烷基、c5-c6杂环基、c2-c12聚乙二醇、o、s、q选自n、c6-c8芳基、c6-c8杂芳基、c3-c8环烷基、c3-c8杂环基;l3'选自聚乙二醇、及其任意组合;x为br;y为r1、r2、r3、r4、r5、r8、r10、r11、r15各自独立地选自h、c1-c4烷基、c6-c8芳基、c6-c8杂芳基、c5-c6环烷基、c5-c6杂环基。进一步的,所述的连接子的结构通式为:或或或优选的,所述的连接子结构可为:本发明还提供了上述连接子在制备抗体药物偶联物中的应用。在本发明的另一方面,提供了一种抗体药物偶联物,所述的抗体药物偶联物的结构如式ii所示:ab-(a-l-d)n(ii)其中,ab是任一抗体或抗体功能性片段。进一步的,抗体包括抗体的衍生物或类似物;“功能性”片段代表所述片段、衍生物或类似物可辨认相同的抗原,辨认衍生自抗原的片段、衍生物或类似物的抗体,例如但不限于:f(ab')2、fab、fab'、fv片段及抗体的重链及轻链二聚体、或其任何最小片段像是fvs或单链抗体(single-chainantibodyfragment/single-chainvariablefragment,scfv)。此外,抗体可为抗体的融合蛋白。抗体也可包括经修饰或未经修饰(亦即,通过任何分子的共价连接)的类似物和衍生物,只要这样的共价连接允许抗体保留其抗原结合免疫特异性。例如但不限于:抗体的类似物和衍生物,包括经过进一步修饰,例如:糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过已知的保护/阻隔基衍生化、蛋白酶切割、连接至细胞抗体单元或其他蛋白质等。可利用已知技术实现任何大量的化学修饰,包括但不限于特异性化学切割、乙酰化、甲酰化、衣霉素存在下的代谢合成等。此外,类似物或衍生物可包括一种或多种非天然氨基酸。在一些实施例中,抗体可在与fc受体作用的氨基酸残基中具有修饰(例如:取代、删除、或增加);更进一步的,所述抗体选自鼠源抗体、哺乳动物来源抗体、嵌合抗体、人源化抗体、人抗体、多特异性抗体。连接子部分包含第一连接子部分a和第二连接子部分l;第一连接子部分a包括与所述ab中巯基或者氨基共价连接的基团。d是药物部分,所述药物包括但不限于细胞毒性药物、细胞分化因子、干细胞营养因子、类固醇类药物、治疗自身免疫疾病的药物、抗炎症药物或传染性疾病的药物。进一步的,所述的药物部分d包括但不限于微管蛋白抑制剂或dna损伤剂。所述微管蛋白抑制剂包括但不限于海兔毒素(dolastatin)及奥瑞他汀(auristatin)类、美登素(maytansine)类;所述dna损伤剂包括但不限于卡奇霉素(calicheamicin)类、倍癌霉素(duocarmycin)类、安曲霉素类衍生物pbd(pyrrolobenzodiazepine,吡咯并苯并二氮杂)、喜树碱类衍生物sn-38、拓扑异构酶i抑制剂。所述奥瑞他汀(auristatin)类药物包括但不限于单甲基澳瑞他汀e(monomethylauristatine;mmae)、单甲基澳瑞他汀f(monomethylauristatinf;mmaf)、和澳瑞他汀f(auristatinf;af)或它们的洐生物,所述美登素类药物包括但不限于dm1、dm3、dm4或它们的洐生物(抗体药物偶联物的弹头分子研究进展,胡馨月等,中国医药生物技术,2017年12月,第12卷第6期)(美登素类抗体药物偶联物研究进展,周磊等,中国新药杂志2016年第25卷第22期,第2521-2530页)。所述药物部分d还可以是瓢菌素(amanitins)、蒽环类物(anthracyclines)、浆果赤霉素(baccatins)、喜树碱(camptothecins)、西马多丁(cemadotins)、秋水仙碱(colchicines)、秋水仙胺(colcimids)、考布他汀(combretastatins)、隐菲辛(cryptophycins)、圆皮海绵内酯(discodermolides)、多烯紫杉醇(docetaxel)、阿霉素(doxorubicin)、棘霉素(echinomycins)、艾榴塞洛素(eleutherobins)、埃博霉素(epothilones)、雌莫司汀(estramustines)、偏端霉素(lexitropsins)、美登素(maytansines)、氨甲蝶呤(methotrexate)、纺锤菌素(netropsins)、嘌呤霉素(puromycins)、根瘤菌素(rhizoxins)、紫杉烷(taxanes)、微管蛋白裂解素(tubulysins)、或长春花生物碱(vincaalkaloids)。所述的药物部分d还可以为维生素a前体、叶酸等。所述药物部分d不仅限于上述类别,还包括所有可用于adc的药物。n为1、2、3、4;其特征在于,所述的第一连接子部分a的结构如式iii所示:其中:m1、m2与抗体或抗体功能性片段部分ab的巯基或者氨基共价连接,其各自独立地选自或者为空,m1和m2可以相同或不同,但不能同时为空;l1、l2各自独立地选自c1-c9烷基、c2-c9烯基、c2-c9炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基、聚乙二醇、o、s、及其任意组合;q选自n、b、p、芳基、杂芳基、环烷基、(杂环基)烷基;l3与第二连接子部分l共价连接,其选自聚乙二醇、o、s及其任意组合或为空;r1为任意的基团或者为空,r2、r3、r4、r5、r9、r10、r11、r12、r13、r14、r15各自独立地选自h、c1-c6烷基、c2-c6烯基、c2-c6炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基;第二连接子部分l为任一可裂解连接子组合或不可裂解连接子或者为空,当l为空时,l3直接与药物部分d共价相连。更进一步的,前述所述的l1、l2、l3、q、r1进一步的分别可为:l1、l2各自独立地选自c1-c4烷基、c6-c8芳基、c6-c8杂芳基、c5-c6环烷基、c5-c6杂环基、c2-c12聚乙二醇、o、s、q选自n、c6-c8芳基、c6-c8杂芳基、c3-c8环烷基、c3-c8杂环基;l3选自聚乙二醇、o、s及其任意组合或为空;r1选自h、c1-c6烷基、c2-c6烯基、c2-c6炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基。更进一步的,所述的第一连接子部分结构通式如下所示:更进一步的,所述的抗体或抗体功能性片段特异性结合细胞表面受体或肿瘤相关抗原。更进一步的,所述的第一连接子部分a包括与所述ab中巯基或者氨基共价连接的基团。更进一步的,所述的第一连接子部分a的结构可为:更进一步的,所述的第二连接子部分l可为:c1-c9烷基、c2-c9烯基、c2-c9炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基、o、s、val-val-pab、val-cit-pab、val-ala-pab、val-lys(ac)-pab、phe-lys-pab、phe-lys(ac)-pab、d-val-leu-lys、gly-gly-arg、ala-ala-asn-pab、ala-pab、pab及其任意组合或为空,其中,r6、r7各自独立地选自h、c1-c6烷基、c2-c6烯基、c2-c6炔基、芳基、杂芳基、c3-c9环烷基、c3-c9杂环基,e为0、1、2、3、4、5、6、7、8。优选的,所述的第二连接子部分l可为:更进一步的,所述的药物部分为细胞毒性药物、细胞分化因子、干细胞营养因子、类固醇类药物、治疗自身免疫疾病的药物、抗炎症药物或传染性疾病的药物。更进一步的,所述的药物部分包括但不限于微管蛋白抑制剂或dna损伤剂。更进一步的,所述的微管蛋白抑制剂包括但不限于海兔毒素(dolastatin)类、奥瑞他汀(auristatin)类、美登素(maytansine)类、;所述dna损伤剂包括但不限于卡奇霉素类(calicheamicin)、倍癌霉素(duocarmycin)类、安曲霉素类衍生物pbd、喜树碱类衍生物(优选的为喜树碱类衍生物sn-38、拓扑异构酶i抑制剂(即dxd))。更进一步的,所述的药物部分包括但不限于瓢菌素(amanitins)、蒽环类物(anthracyclines)、浆果赤霉素(baccatins)、喜碱(camptothecins)、西马多丁(cemadotins)、秋水仙碱(colchicines)、秋水树仙胺(colcimids)、考布他汀(combretastatins)、隐菲辛(cryptophycins)、圆皮海绵内酯(discodermolides)、多烯紫杉醇(docetaxel)、阿霉素(doxorubicin)、棘霉素(echinomycins)、艾榴塞洛素(eleutherobins)、埃博霉素(epothilones)、雌莫司汀(estramustines)、偏端霉素(lexitropsins)、美登素(maytansines)、氨甲蝶呤(methotrexate)、纺锤菌素(netropsins)、嘌呤霉素(puromycins)、根瘤菌素(rhizoxins)、紫杉烷(taxanes)、微管蛋白裂解素(tubulysins)、或长春花生物碱(vincaalkaloids)、、维生素a前体、叶酸、喜树碱类衍生物sn-38、拓扑异构酶i抑制剂(即dxd)。优选的,所述的药物部分d可为:在某些优选的实施例中,所述的抗体药物偶联物的结构为:adc1adc2adc3adc4adc5adc6adc7adc8adc9adc10adc11adc12adc13adc14adc15adc16其中:所述的抗体或抗体功能性片段ab是针对细胞表面受体和肿瘤相关抗原的抗体;n1、n2、n3独立的为0、1、2、3、4,,n1、n2、n3不同时为0,且n1+n2+n3≤4;n4、n5、n6、n7独立的为0、1、2、3、4,n4、n5、n6、n7不同时为0,且n4+n5+n6+n7≤4;n8、n9独立的为0、1、2、3、4,n8、n9不同时为0,且n8+n9≤4;m1、m2、m3独立的为0、1、2,m1、m2、m3至少一个为0,但m1、m2、m3不同时为0,且m1+m2+m3≤2;m4、m5独立的为0、1、2,m4、m5不同时为0,且m4+m5≤2。进一步的,所述抗体药物偶联物的结构为:adc17adc18adc19adc20adc21adc22adc23adc24adc25adc26adc27adc28adc29adc30adc31adc32其中:所述ab是针对细胞表面受体和肿瘤相关抗原的抗体;n10为0、1、2、3、4;m6为0、1、2。进一步的,所述抗体药物偶联物的结构为:adc33adc34adc35adc36adc37adc38adc39adc40adc41adc42adc43adc44adc45adc46adc47adc48其中:所述ab是针对细胞表面受体和肿瘤相关抗原的抗体;n11为0、1、2、3、4;m7为0、1、2。本发明还提供了一种药物组合物,其含有如本发明所述的任一抗体药物偶联物以及药学上可接受的载体。本发明还提供了一种利用本发明所述的任一连接子在制备抗体药物偶联物中的用途。本发明还提供了根据本发明所述的任一抗体药物偶联物,或根据本发明所述的任一药物组合物,在制备治疗癌症、自身免疫性疾病、炎症性疾病或传染性疾病的药物中的用途。附图说明图1a为抗体药物偶联物her2-a'-7-val-cit-pab-mmaehic-hplc色谱图(偶联时加入的药物与抗体巯基的摩尔比为5:1)。图1b为抗体药物偶联物her2-a'-11-val-cit-pab-mmaehic-hplc色谱图(偶联时加入的药物与抗体巯基的摩尔比为5:1)。图1c为抗体药物偶联物her2-a'-10-val-cit-pab-mmaehic-hplc色谱图(偶联时加入的药物与抗体巯基的摩尔比为5:1)。图1d为抗体药物偶联物her2-575dz-val-cit-pab-mmaehic-hplc色谱图(偶联时加入的药物与抗体巯基的摩尔比为5:1)。图2a给出了her2-a'-11-val-cit-pab-mmae(偶联缓冲液ph分别为8.5、9.0、7.4)、her2-a'-7-val-cit-pab-mmae(偶联缓冲液ph分别为8.5、9.0、7.4)、her2-a'-10-val-cit-pab-mmae(偶联缓冲液ph分别为8.5、9.0、7.4)(偶联缓冲液ph为9.0)的sds-page电泳图谱。图2b给出了her2-a'-7-val-cit-pab-mmae、her2-575dz-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae的sds-page电泳图谱(偶联缓冲液ph为9.0)。图3采用elisa法测定抗体药物偶联物(其中抗体药物偶联物为her2-a'-7-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae,her2抗体和her2-mc-val-cit-pab-mmae为阴性和阳性对照)对抗原的亲和力曲线图,其中浓度为横坐标,光密度吸收值为纵坐标。图4a抗体药物偶联物(her2-a'-11-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae)对人乳腺癌sk-br-3细胞的增殖抑制率曲线图。图4b抗体药物偶联物(her2-a'-8-val-cit-pab-mmae、her2-a'-7-val-cit-pab-mmae)对人乳腺癌sk-br-3细胞的增殖抑制率曲线图。图5给出了抗体(ab)与第一连接子部分(a)偶联物(ab-a)的偶联结果,编号1、2、3为her2-a'-7,偶联缓冲液ph分别为8.5、9.0、7.4;编号4、5、6为her2-a'-8,偶联缓冲液ph分别为8.5、7.4、9.0;编号7、8、9为her2-a'-10,偶联缓冲液ph分别为8.5、9.0、7.4;编号10、11、12为her2-a'-11,偶联缓冲液ph分别为9.0、7.4、9.5;编号13、14、15为her2-a'-12,偶联缓冲液ph分别为7.4、9.0、8.5;编号16、17、18为her2-a'-20,偶联缓冲液ph分别为8.5、9.0、7.4;编号19、20、21为her2-a'-28,偶联缓冲液ph分别为9.0、8.5、7.4;编号22、23、24为her2-a'-31,偶联缓冲液ph分别为7.4、8.5、9.0。图6a是裸抗体(claudine18.2抗体)的lc-ms图谱。图6b是claudine18.2-a’-7-vc-pab-mmae的lc-ms图谱。图7a是裸抗体(claudine18.2抗体)的等电点分布图谱。图7b是claudine18.2-a’-7-vc-pab-mmae的等电点分布图谱。具体实施方式下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。[定义]除非另有定义,本文使用的所有科技术语具有本领域普通技术人员所理解的相同含义。尽管本发明的广义范围所示的数字范围和参数近似值,但是具体实施例中所示的数值尽可能准确的进行记载。然而,任何数值本来就必然含有一定的误差,其是由它们各自的测量中存在的标准偏差所致。另外,本文公开的所有范围应理解为涵盖其中包含的任何和所有子范围。例如记载的“1至10”的范围应认为包含最小值1和最大值10之间(包含端点)的任何和所有子范围;也就是说,所有以最小值1或更大起始的子范围,例如1至6.1,以及以最大值10或更小终止的子范围,例如5.5至10。另外,任何称为“并入本文”的参考文献应理解为以其整体并入。本发明中所用的“”是指含“”的基团在此处与其他基团通过化学键连接。本发明中的术语“抗体”以最广义使用,并且涵盖各种抗体结构,包括但不限于单克隆抗体、多克隆抗体、多特异性抗体(例如双特异性抗体)、和抗体片段。特别的,本文所使用的“抗体”指包含通过二硫键而互连的至少两条重(h)链和两条轻(l)链的蛋白。每条重链包含一重链可变区(缩写为vh)和一重链恒定区。该重链恒定区包含三个域(domain),ch1、ch2和ch3。每条轻链包含一轻链可变区(缩写为vl)和一轻链恒定区。该轻链恒定区包含一个域,cl。vh和vl区域还可再细分为具有高可变性的多个区,被称为互补决定区(cdr),其间散布有更为保守的被称为框架区(fr)的多个区域。每个vh和vl均由三个cdr和四个fr构成,按照以下顺序从氨基端至羧基端排布:fr1、cdr1、fr2、cdr2、fr3、cdr3、fr4。重链和轻链的这些可变区包含与抗原相互作用的结合域。抗体的恒定区可介导免疫球蛋白与宿主的组织或因子结合,包括免疫系统的各种细胞(如效应细胞)和经典补体系统的第一成分(clq)。嵌合或人源化抗体也涵盖在根据本发明的抗体中。术语“人源化抗体”是指一种抗体,其包含来源于非人源抗体的cdr区、并且该抗体分子的其他部分来源于一种(或几种)人抗体。而且,为了保留结合亲和力,可以修饰骨架(称为fr)区段的一些残基(jonesetal.,nature,321:522-525,1986;verhoeyenetal.,science,239:1534-1536,1988;riechmannetal.,nature,332:323-327,1988)。通过本领域技术人员已知的技术可以制备根据本发明的人源化抗体或其片段(例如,描述于文件singeretal.,j.immun.150:2844-2857,1992;mountainetal.,biotechnol.genet.eng.rev.,10:1-142,1992;或bebbingtonetal.,bio/technology,10:169-175,1992)。术语“嵌合抗体”是指以下抗体,其中的可变区序列来自一物种而恒定区序列来自另一物种,例如可变区序列来自小鼠抗体而恒定区序列来自人抗体的抗体。通过使用基因重组技术可以制备根据本发明的嵌合抗体或其片段。例如,所述嵌合抗体可以通过克隆重组dna来生产,所述重组dna包含启动子和编码根据本发明的非人尤其是鼠单克隆抗体可变区的序列、以及编码人抗体恒定区的序列。由这种重组基因编码的本发明嵌合抗体将是,例如,鼠-人嵌合体,该抗体的特异性由来源于鼠dna的可变区确定,并且其同种型由来源于人dna的恒定区来确定。对于制备嵌合抗体的方法,例如,可以参考文件verhoeynetal.(bioessays,8:74,1988)。术语“单克隆抗体”系指具有单一分子组成的抗体分子的制备物。单克隆抗体组合物显示出对于特定表位的单一结合特异性和亲和性。本发明中的术语“功能性片段”是指抗体片段由其来源抗体的重或轻可变链的部分序列构成或者包含它们,所述部分序列足以保留与其来源抗体相同的结合特异性和充分的亲和力,优选至少等于其来源抗体亲和力的1/100,在更优选方式中至少等于1/10。这种功能片段将包含最少5个氨基酸,优选其来源的抗体序列的10、15、25、50和100个连续氨基酸。本发明中的术语“dar”又称药物抗体偶联比率(drugtoantibodyratio),它是抗体药物偶联物的一个独有的质量属性。高的dar值可能会影响adc的安全性,且其治疗窗口较窄。低的dar值可能会导致疗效的下降,但是其安全性强,且具有较宽的治疗窗口。最佳的平均dar值范围为2-4。本发明中的术语“连接子”是指一种能作为“桥梁”,分别与抗体/抗体功能性片段和药物发生反应,将两者连接起来的化合物。本发明涉及的“连接子”包括第一连接子部分和第二连接子部分,本发明中的术语“第一连接子部分”包括两个相同或者不同的连接基团,该连接基团能同时与抗体/抗体功能性片段上的链间巯基或者氨基共价连接,所述的连接基团为任意能与巯基或者氨基共价连接的官能团。本发明中的术语“第二连接子部分”用于与药物偶联,其为任意的可裂解或者不可裂解的连接子,可裂解连接子依赖于胞内过程来释放毒素,如胞质内还原过程、暴露于溶酶体内酸性条件或被细胞内特异性蛋白酶所裂解。不可裂解的连接子需要在adc抗体部分的蛋白降解之后,方可释放出药物。本发明中的术语“药物”泛指任何具有期望的生物活性,并具有反应性官能团以便制备本发明所述偶联物的化合物。期望的生物活性包括诊断、治愈、缓解、治疗、预防人或其它动物的疾病。随着新型药物不断被发现和发展,这些新药也应纳入本发明所述的药物中。具体的,所述的药物包括但不限于细胞毒性药物、细胞分化因子、干细胞营养因子、类固醇类药物、治疗自身免疫疾病的药物、抗炎症药物或传染性疾病的药物。更具体的,所述的药物包括但不限于微管蛋白抑制剂或者dna、rna损伤剂。在本发明的一些实施方案中,所述药物选自:美坦生类化合物、v-atp酶抑制剂、促凋亡剂、be12抑制剂、mcl1抑制剂、hsp90抑制剂、iap抑制剂、mtor抑制剂、微管稳定剂、微管去稳定剂、阿里他汀(auristatin)、多拉司他汀(dolastatin)、metap(甲硫氨酸氨肽酶)、蛋白质crml的核输出抑制剂、dppiv抑制剂、蛋白酶体抑制剂、线粒体中磷酰基转移反应的抑制剂、蛋白质合成抑制剂、激酶抑制剂、cdk2抑制剂、cdk9抑制剂、驱动蛋白抑制剂、hdac抑制剂、dna损伤剂、dna烷基化剂、dna嵌入剂、dna小沟结合剂、dhfr抑制剂、以及海兔毒素肽、维生素a前体、叶酸、喜树碱类衍生物。在本发明的一些优选实施方案中,所述药物为细胞毒性药物(例如抗代谢药、抗肿瘤抗生素、生物碱)、免疫增强剂或放射性同位素。优选地,所述药物可选自瓢菌素(amanitins)、蒽环类物(anthracyclines)、浆果赤霉素(baccatins)、喜碱(camptothecins)、西马多丁(cemadotins)、秋水仙碱(colchicines)、秋水树仙胺(colcimids)、考布他汀(combretastatins)、隐菲辛(cryptophycins)、圆皮海绵内酯(discodermolides)、多烯紫杉醇(docetaxel)、阿霉素(doxorubicin)、棘霉素(echinomycins)、艾榴塞洛素(eleutherobins)、埃博霉素(epothilones)、雌莫司汀(estramustines)、偏端霉素(lexitropsins)、美登素(maytansines)、氨甲蝶呤(methotrexate)、纺锤菌素(netropsins)、嘌呤霉素(puromycins)、根瘤菌素(rhizoxins)、紫杉烷(taxanes)、微管蛋白裂解素(tubulysins)、或长春花生物碱(vincaalkaloids)。更优选地,所述“药物”可选自mmad(单甲基澳瑞他汀d,monomethylauristatind)及其衍生物、mmae(单甲基澳瑞他汀e,monomethylauristatine)及其衍生物、mmaf(单甲基澳瑞他汀f,monomethylauristatinf)及其衍生物、美登素类衍生物dm1(mertansinederivativem1)、美登素类衍生物dm4(mertansinederivativem4)、倍癌霉素(duocarmycine)及其衍生物、卡其霉素(calicheamicin)及其衍生物、pbda(pyrrolobenzodiazepines)、阿霉素(doxorubicin)、长春花生物碱(vincaalkaloids)、metrotrexate、长春花碱(vinblastine)、道诺霉素(daunorubicin)及其衍生物、微管蛋白裂解素(tubulysins)及其衍生物、喜树碱类衍生物sn-38、拓扑异构酶i抑制剂(即dxd)。在一些具体的实施例中,所述的“药物”可以为以下物质及其衍生物:①美登素:②类美登素类衍生物:③安丝菌素衍生物:④丙氨酰基美登醇:⑤mmae:⑥mmad:⑦mmaf:⑧tubulysind:⑨卡其霉素:⑩阿霉素:吡咯并苯二氮卓类衍生物:维生素a前体:叶酸:拓扑异构酶i抑制剂(即dxd):本发明中的术语“抗体药物偶联物”表示抗体/抗体功能性片段、连接子(第一连接子部分、第二连接子部分)、药物部分经过化学反应连接在一起的化合物,本发明涉及的抗体药物偶联物尽管仍旧是混合物,但是与传统方式得到的偶联物相比,其dar分布范围很窄,最佳抗体药物偶联物平均dar值范围为2-4。本发明提供的抗体药物偶联物在制备时,第一连接子部分(a)与第二连接子部分-药物组合物(l-d)组合物(a-l-d)在与抗体(如本实施例提供的claudin18.2抗体)进行偶联时,在易水解条件下可以发生水解,其水解部位在第一连接子的马来酰亚胺部分,以ab--a’-7-vc-pab-mmae(且一个抗体偶联4个药物的情况下)为例,当ab--a’-7-vc-pab-mmae偶联物发生4个水解时,其结构示意图为:或者当ab--a’-7-vc-pab-mmae偶联物发生7个水解时,其结构示意图为:当ab--a’-7-vc-pab-mmae偶联物发生8个水解时,其结构示意图为:本发明中的术语“药物组合物”表示组合在一起以实现某种特定目的的至少一种药物以及任选地可药用载体或辅料的组合。在某些实施方案中,所述药物组合物包括在时间和/或空间上分开的组合,只要其能够共同作用以实现本发明的目的。例如,所述药物组合物中所含的成分可以以整体施用于对象,或者分开施用于对象。当所述药物组合物中所含的成分分开地施用于对象时,所述成分可以同时或依次施用于对象。优选地,所述可药用载体是水、缓冲水溶液、等渗盐溶液如pbs(磷酸盐缓冲液)、葡萄糖、甘露醇、右旋葡萄糖、乳糖、淀粉、硬脂酸镁、纤维素、碳酸镁、0.3%甘油、透明质酸、乙醇或聚亚烷基二醇如聚丙二醇、甘油三酯等。所用可药用载体的类型尤其依赖于根据本发明的组合物是否配制为用于口服、鼻、皮内、皮下、肌内或静脉施用。根据本发明的组合物可包含润湿剂、乳化剂或缓冲液物质作为添加剂。根据本发明的药物组合物、疫苗或者药物制剂可通过任何适宜的途径施用,例如可口服、鼻、皮内、皮下、肌内或静脉内施用。本发明中的术语“烷基”表示直链或者支链的饱和碳氢化合物(即,不包含双键或三键)。烷基基团可以带有1到9个碳原子(当其在本发明中出现时,“1到9”的数值范围表示此范围中的任意一个整数,例如,“1到9个碳原子”表示此烷基基团可以包含1个碳原子、2个碳原子、3个碳原子等,一直到9个碳原子。同时,此烷基的定义也包括无指定链长度的烷基)。烷基基团可以是包含1到9个碳原子的中等尺寸烷基。典型的烷基基团包括但不限于:甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基等。本发明中的术语“烯基”表示包含一个或多个双键的直链或者支链的碳氢化合物。烯基基团可以带有2到9个碳原子,也包括无指定链长度的烯基。烯基基团可以是包含2到9个碳原子的中等尺寸烯基。烯基基团也可以是包含2到4个碳原子的小尺寸烯基。烯基基团可以被设计为“c2-4烯基”或者相似设计。仅举例而言,“c2-4烯基”表示烯基链中有2-4个碳原子,即,烯基链的选择范围如下:乙烯基、丙烯-1-基、丙烯-2-基、丙烯-3-基、丁烯-1-基、丁烯-2-基、丁烯-3-基、丁烯-4-基、1-甲基-丙烯-1-基、2-甲基-丙烯-1-基、1-乙基-乙烯-1-基、2-甲基-丙烯-3-基、丁-1,3-二烯基、丁-1,2-二烯基、丁-1,2-二烯基-4-基。典型的烯基包括,但不限于:乙烯基、丙烯基、丁烯基、戊烯基和己烯基等。本发明中的术语“炔基”表示包含一个或多个三键的直链或者支链的碳氢化合物。炔基基团可以带有2到9个碳原子,也包括无指定链长度的炔基。炔基基团可以是包含2到9个碳原子的中等尺寸炔基。炔基基团可也以是包含2到4个碳原子的低级炔基。炔基基团可以被设计为“c2-4炔基”或者相似设计。仅举例而言,“c2-4炔基”表示炔基链中有2-4个碳原子,即,炔基链可选自:乙炔基、丙炔-1-基、丙炔-2-基、丁炔-1-基、丁炔-2-基、丁炔-3-基和2-丁炔基。典型的炔基包括但不限于:乙炔基、丙炔基、丁炔基、戊炔基和己炔基等。本发明中的术语“芳基”表示具有共轭π电子系统的环或环系统,并且包括碳环芳基(例如苯基)和杂环芳基(例如:吡啶)。该术语包括单环或多个稠环(即共享一对相邻原子的环)基团,整个环系统是芳香性的。本发明中的“杂芳基”表示包含一个或多个杂原子的芳香环或环体系,(即,共用两个相邻原子的两个或多个稠环),即在环骨架上除碳外,包括但不限于,氮、氧、硫等元素。当杂芳基是一个环系统时,系统中的每个环都是芳香族的。杂芳基可具有5-18个环成员(即,构成环骨架的原子数,包括碳原子和杂原子的数目),目前的定义也包括无指定环大小的杂芳基。杂芳基的实例包括但不限于,呋喃基、噻吩基、酞嗪基、吡咯基、恶唑基、噻唑基、咪唑基、吡唑基、异恶唑基、异噻唑基、三唑、噻二唑基、吡啶基、哒嗪基、嘧啶基、吡嗪基、三嗪基、喹啉基、异喹啉基、苯并咪唑基、苯并恶唑基、苯并噻唑基、吲哚基、异吲哚基、苯并噻吩基。本发明中的“环烷基”表示完全饱和的碳环或环体系。实例包括但不限于,环丙基、环丁基、环戊基、环己基。本发明中的“(杂环基)烷基”是指杂环基作为取代基通过亚烷基与其他基团相连。实例包括但不限于,咪唑啉甲基和二氢吲哚基乙基。所述的“杂环基”是指在非芳香环或环体系的骨架中含有至少一个杂原子。杂环基可以稠环、桥环或螺环的方式连接。杂环基的环体系中至少有一个环是非芳香性的,其可具有任意的饱和度。所述的杂原子可位于该环体系的非芳香环或芳香环上。所述杂环基可以具有3至20个环上原子(即,构成环骨架的原子数,包括碳原子和杂原子的数目),目前的定义也包括无指定环数目范围的杂环基。杂环基团可以是包含3到10个环上原子的中等尺寸杂环基,杂环基团可以是包含3到10个环上原子的中等尺寸杂环基。杂环基团也可以是包含3到6个环上原子的小尺寸杂环基。杂环基的实例包括但不限于:氮杂环庚烯基、吖啶基、咔唑基、噌啉基、二氧戊环基、咪唑啉基、咪唑烷基、吗啉基、环氧乙烷基、氧杂环庚烷基、硫杂环丁烷基、哌啶基、哌嗪基、吡唑啉基、吡唑烷基、1,3-二氧杂环己烯基、1,3-二恶烷基、1,4-二氧杂环己烯基、1,4-二恶烷基、1,3-氧硫杂环己烷基、1,4-氧硫杂环己烷基、1,4-氧硫杂环己烷基、2h-1,3-二氧戊环基、1,3-二硫戊环基、1,3-二硫戊环基、异恶唑啉基、异恶唑烷基、恶唑啉基、恶唑烷基、恶唑烷酮基、恶唑烷酮基、噻唑烷基、1,3-氧硫杂环戊烯基、二氢吲哚基、异吲哚啉基、四氢呋喃基、四氢吡喃基、四氢噻吩基、四氢噻喃基、四氢-1,4-噻嗪基、硫代吗啉基、二氢苯并呋喃基、苯并咪唑烷基和四氢喹啉。[具体实施例]通用步骤a:溴代马来酰亚胺连接子的合成通用方法将胺类及其盐或boc保护衍生物(1eq)及无水乙酸钠(2.5eq)溶于乙酸中(1mmol/2ml),升温至50-100℃,随后快速加入溴马来酸酐(2.5eq),50-100℃反应过夜;降温至45℃,减压浓缩,剩余物中加入乙酸乙酯,洗涤,干燥,浓缩得粗品。粗品经正向柱层析或反向制备液相纯化得到目标产物。通用步骤b:苯硫酚马来酰亚胺连接子的合成通用方法将溴马来酰亚胺连接子(1.0eq)溶于无水dmf(1mmol/5ml)中,加入吡啶(3.0eq),室温搅拌5-10分钟。将苯硫酚钠(2.1eq)溶于无水dmf(1mmol/1ml)中,滴加到反应液中,滴加完毕室温搅拌0.5-5h。反应完成后,加入5倍体积的蒸馏水,析出固体,过滤,洗涤,干燥,浓缩得粗品。粗品经正向柱层析或反向制备液相纯化得到目标产物。实施例1a'-1的合成使用通用步骤a,加入化合物1(即3,5-二氨基苯甲酸)300mg,其他按摩尔比加入。70℃下反应过夜。经制备液相纯化,得到目标化合物a'-1。lc-ms(esi+)468.9[m+h]+。实施例2a'-4的合成使用通用步骤b,加入原料a'-1100mg,其他按摩尔比加入。经制备液相纯化,得到目标化合物a'-4。lc-ms(esi+)529.0[m+h]+。实施例3a'-2的合成化合物2(1个摩尔比)溶解于乙醇、水的混合溶液中,搅拌,一次性加入还原铁粉(5个摩尔比),氯化铵(3个摩尔比),80℃反应1小时。反应结束后,冷却到室温,过滤,滤去固体,减压蒸除溶剂,得到化合物3,lc-msm/z(es+),154.05(m+h)+。使用通用步骤a,加入化合物3(1个摩尔比),其他按摩尔比加入。80℃下反应过夜。经制备液相纯化,得到目标化合物a'-2,lc-msm/z(es+),471.85(m+h)+。实施例4a'-7的合成将化合物5(boc-咪唑,2.37g,14.1mmol)用30ml甲苯溶解,室温加入化合物4(二亚乙基三胺,0.66g,6.41mmol),60℃加热搅拌6h。反应结束后,减压蒸馏得到油状粗品,粗品用40ml二氯甲烷溶解,水洗(40ml×3),干燥,过滤,浓缩,得1.84g化合物6,收率95%。lc-ms(esi+)304.2[m+h]+。将化合物6(3.3g,10.89mmol)溶于30ml无水甲苯中,加入化合物7(丁二酸酐,1.3g,13mmol),60℃加热搅拌过夜。反应完成后,减压蒸馏除去甲苯,残余物溶于20ml二氯甲烷中,水洗(20ml×2),饱和nacl洗(20ml×1),干燥,过滤,减压蒸馏,得3.5g化合物8,收率80%。lc-ms(esi+)404.3[m+h]+。使用通用步骤a,加入原料8100mg,其他按摩尔比加入。70℃下反应过夜。经制备液相纯化,得到目标化合物a'-725.8mg,收率20%。lc-ms(esi+)519.9[m+h]+。实施例5a'-8的合成向化合物9(1,3-二氨基丙醇,5.0g,55.47mmol)中加入50mlthf和20ml水,搅拌溶解。室温下滴加boc2o(25.4g,116.5mmol,2.1eq)的30mlthf溶液。滴加完毕后,室温搅拌过夜。反应结束后,减压浓缩,残余物加入30ml石油醚,室温搅拌20min,有白色固体析出,过滤,干燥,得白色化合物1014.7g,收率91%,lc-ms(esi+)291.2[m+h]+。向化合物10(14.7g,50.64mmol)中加入100ml无水thf,搅拌溶解,冰浴中降温。随后分批加入nah(4.8g,202.55mmol,4eq),搅拌10min。滴加化合物11(溴乙酸叔丁酯,24.7g,126.6mmol,2.5eq)的50mlthf溶液,滴加完毕,升温至40℃搅拌过夜。反应结束后,降至室温,用250ml乙酸乙酯稀释,缓慢加入冰水混合物直至无大量气泡生成,分液,水相用乙酸乙酯萃取,合并有机相,洗涤,干燥,浓缩,残余物加入50ml石油醚室温搅拌20min,析出白色固体,过滤,干燥得白色化合物1212.1g,收率60%,lc-ms(esi+)404.1[m+h]+。向化合物12(12.1g,30.0mmol)中加入120ml无水dcm,搅拌溶解,再加入氯化氢的1,4-二氧六环溶液(4m,45ml,180mmol),室温搅拌过夜。反应完毕,减压浓缩,残余物用30ml甲基叔丁基醚超声分散,离心,弃去清液,再加入30ml石油醚分散,离心,弃去清液。固体经减压蒸馏除去溶剂,得白色化合物137.7g,收率100%,lc-ms(esi+)148.1[m+h]+。使用通用步骤a,加入原料131.2g,其他按摩尔比加入。85℃下反应5h。经制备液相纯化,得到目标化合物a'-8890mg,收率35%。lc-ms(esi+)464.9[m+h]+。实施例6a'-9的合成使用通用步骤a,加入原料化合物14(2,3-二氨基丙酸)500mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-9240mg,收率23%。lc-ms(esi+)420.9[m+h]+。实施例7a'-10的合成使用通用步骤b,加入原料a'-7104mg,其他按摩尔比加入。室温下反应2.5h。经制备液相纯化,得到目标化合物a'-1027.7mg,收率24%。lc-ms(esi+)596.1[m+h]+。实施例8a'-11的合成使用通用步骤b,加入原料a'-8600mg,其他按摩尔比加入。室温下反应2.5h。经制备液相纯化,得到目标化合物a'-11203mg,收率30%。lc-ms(esi+)525.1[m+h]+。实施例9a'-12的合成使用通用步骤b,加入原料a'-9120mg,其他按摩尔比加入。室温下反应0.7h。经制备液相纯化,得到目标化合物a'-1230mg,收率22%。lc-ms(esi+)481.0[m+h]+。实施例10a'-13的合成使用通用步骤a,加入化合物15(l-赖氨酸)500mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-13410mg,收率26%。lc-ms(esi+)462.9[m+h]+。实施例11a'-14的合成向化合物16(500mg,2.03mmol)中加入1ml四氢呋喃和1ml饱和nahco3水溶液,搅拌溶解,冰浴搅拌5min,分批缓慢加入化合物17(724.4mg,4.67mmol),冰浴搅拌40min,转移至室温搅拌2.5h。反应结束后,旋蒸除去thf,加入1ml甲醇稀释,制备液相分离得化合物18245mg,收率37%。lc-ms(esi+)327.1[m+h]+。使用通用步骤a,加入原料18245mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-1467mg,收率23%。lc-ms(esi+)462.9[m+h]+。实施例12a'-15的合成使用通用步骤a,加入化合物19(boc-4-氨基-l-苯丙氨酸)56.1mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-1517.9mg,收率18%。lc-ms(esi+)496.9[m+h]+。实施例13a'-16的合成使用通用步骤b,加入原料a'-13218mg,其他按摩尔比加入。室温下反应0.7h。经制备液相纯化,得到目标化合物a'-1665mg,收率26%。lc-ms(esi+)523.1[m+h]+。实施例14a'-19的合成向化合物20(140mg,0.5mmol)加入3ml二氯甲烷溶解,加入三乙胺(210μl,1.5mmol),冰浴搅拌,将化合物21(62.5μl,0.75mmol)溶于2mldcm,滴加至反应中,冰浴搅拌0.5h,室温搅拌2h。反应结束后,反应液浓缩,残余物用乙腈溶解,制备液相分离得2293.5mg,收率56.1%。lc-ms(esi+)335.2[m+h]+。使用通用步骤a,加入原料2233.4mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-197.5mg,收率19.1%。lc-ms(esi+)393.0[m+h]+。实施例15a'-20的合成将23(230mg,1mmol)溶于3ml四氢呋喃中,加入溴马来酸酐(354mg,2mmol),室温搅拌3h。反应结束后,旋蒸除去四氢呋喃,得化合物24粗品。将化合物24用3ml乙酸酐溶解,加入乙酸钠(41mg,0.5mmol),70℃搅拌8h。反应结束后,旋蒸除去溶剂,残余物用4ml乙腈/水(1:1)溶解,经制备液相分离纯化得到化合物2570mg,收率18%。lc-ms(esi+)389.0[m+h]+。向化合物25(99.2mg,0.32mmol)中加入2ml氯化氢1,4-二氧六环溶液(4m,8mmol),冰浴搅拌2h。反应结束后,旋蒸除去溶剂,残余物用乙醚洗(5ml×3),干燥得化合物2664.1mg,收率95.3%。lc-ms(esi+)289.0[m+h]+。向化合物26(21mg,0.1mmol)加入1ml二氯甲烷溶解,加入三乙胺(42μl,0.3mmol),冰浴搅拌,将化合物21(12.5μl,0.15mmol)溶于1mldcm,滴加至反应中,冰浴搅拌0.5h,室温搅拌2h。反应结束后,反应液浓缩,残余物用乙腈溶解,制备液相分离得a'-2018.2mg,收率69.1%。lc-ms(esi+)343.0[m+h]+。实施例16a'-26的合成将化合物27(150mg,0.63mmol,1.0eq)溶解于5ml无水thf中,而后在ar保护下降温至-5℃,小心加入bh3-thf(1n,2.5ml,2.5mmol,4.0eq);搅拌10min,移至室温搅拌10min,而后平稳升温至65℃反应过夜。反应结束后,将反应液降温至0℃,缓慢将5ml甲醇滴加入反应液中,搅拌30min,移至室温搅拌10min,升温至60℃搅拌3h,减压浓缩干,得化合物28粗品,140mg油状液体,不进行纯化,直接投入下一步,lc-ms(esi+)167.1[m+h]+。将化合物28粗品(140.0mg,0.58mmol,1.0eq)溶解于5mlthf中,加入3ml水,而后滴加boc2o(255.5g,1.17mmol,2.0eq)的2.5mlthf溶液,室温搅拌过夜。反应结束后,减压浓缩,残余物中加入30mlea,洗涤,干燥,浓缩,残余物经纯化得化合物29,185mg白色固体,收率72%,lc-ms(esi+)367.1[m+h]+。将化合物29(94.0mg,0.25mmol,1.0eq)溶解于3mldmf中,加入k2co3(71.0mg,0.51mmol,2.0eq),而后滴加化合物11(75.0mg,0.38mmol,1.5eq)的1mldmf溶液,升温至55℃搅拌过夜。反应结束后,反应液中加入20mlea,用30%柠檬酸溶液调节ph=3,萃取,洗涤,干燥,浓缩得化合物30,108mg类白色固体,收率88%,lc-ms(esi+)481.3[m+h]+。将化合物30(108mg,0.22mmol,1.0eq)溶于5ml无水dcm中,再加入hcl的1,4-二氧六环溶液(4m,1.5ml,6.0mmol,27.0eq),室温搅拌3h。反应结束后,减压浓缩,用10ml甲基叔丁基醚超声分散,离心,弃去清液,再加入10ml石油醚分散,离心,弃去清液。固体经减压蒸馏除去溶剂,得化合物31,66.6mg白色固体,收率100%,lc-ms(esi+)225.1[m+h]+。使用通用步骤a,加入化合物3166.6mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-2640mg,收率33%。lc-ms(esi+)540.9[m+h]+。实施例17a'-27的合成化合物a'-26(40.0mg,0.074mmol,1.0eq),用1.0ml无水thf溶解,加入tea(22.5mg,0.22mmol,3.0eq)的500μlthf溶液,室温搅拌5分钟;将化合物32(20.5mg,0.16mmol,2.2eq)溶于500μldmf中滴加于反应液中,滴加完毕室温搅拌过夜。反应结束后,将15ml乙酸乙酯加入反应液中,15%柠檬酸溶液洗涤,水洗,饱和nacl溶液洗,干燥,浓缩,纯化得化合物a'-27,12mg白色固体,收率26%,lc-ms(esi+)629.1[m+h]+。实施例18a'-28的合成将化合物33(150mg,2mmol,1.0eq)溶解于5mldioxane中,加入3ml饱和碳酸氢钠水溶液,而后滴加boc2o(523mg,2.40mmol,1.2eq)的2.5mldioxane溶液,室温搅拌2h;减压浓缩,剩余物中加入30mlea,用30%柠檬酸调节ph=3,萃取,洗涤,干燥,浓缩,加入15mlpe分散,过滤,干燥得344mg化合物34,收率62%,lc-ms(esi+)176.1[m+h]+。将化合物34(350mg,2.00mmol,1.0eq)溶解于4mldmf中,ar气氛下降温至-15℃,加入tea(606.0mg,6.00mmol,3.0eq)的1mldmf溶液,搅拌5min,而后加入t3p(700.0mg,2.20mmol,1.1eq)的1mldmf溶液,搅拌1.5h;将化合物35(135.0mg,1.00mmol,0.5eq)的1mldmf溶液滴加入反应液中,搅拌过夜;加入30mlea,用30%柠檬酸调节ph=3,萃取,洗涤,干燥,浓缩得350mg油状液体化合物36,不纯化直接投入下一步,lc-ms(esi+)449.2[m+h]+。化合物36(350mg,0.78mmol,1.0eq),加入5ml无水dcm搅拌溶解,再加入hcl的1,4-二氧六环溶液(4m,2.0ml,8.0mmol,10.2eq),室温搅拌2h;减压浓缩,用20ml甲基叔丁基醚超声分散,离心,弃去清液,再加入20ml石油醚分散,离心,弃去清液。固体经减压蒸馏除去溶剂,得化合物37254mg,收率100%,lc-ms(esi+)249.1[m+h]+。使用通用步骤a,加入化合物37195mg,其他按摩尔比加入。70℃下反应过夜。经正向柱层析纯化,得到目标化合物a'-2858mg,淡黄色固体,收率17%。lc-ms(esi+)564.9[m+h]+。实施例19a'-29的合成使用通用步骤b,加入化合物3858mg,其他按摩尔比加入。室温下反应0.7h。经正向柱层析纯化,得到目标化合物a'-2914.1mg,收率22%。lc-ms(esi+)625.1[m+h]+。实施例20a'-31的合成将化合物43(342.0mg,3.00mmol,1.0eq)用15ml无水thf溶解,滴加入溴马来酸酐(1110.0mg,6.30mmol,2.1eq),室温搅拌过夜。反应完成后,过滤,滤饼用dcm洗涤,干燥得化合物44,1160mg白色固体,收率83%,lc-ms(esi+)466.9[m+h]+。向化合物45(300mg,0.64mmol,1.0eq)及无水乙酸钠(158.0mg,1.93mmol,3.0eq)中加入6ml乙酸,升温至85℃反应过夜;降温至45℃,减压浓缩,剩余物中加入乙腈20ml,过滤,滤液制备液相纯化得15mg淡黄色固体(即化合物a'-31),收率5%。lc-ms(esi+)430.9[m+h]+。实施例21a'-33的合成使用通用步骤a,加入化合物48(95mg,0.5mmol)和化合物8(80.6mg,0.2mmol),得到化合物a'-3310.9mg,收率10.1%。lc-ms(esi+)548.0[m+h]+。实施例22a'-34的合成使用通用步骤a,加入化合物49(2-氨基-6-氯异烟酸,86mg,0.5mmol),得到化合物a'-3421.3mg,收率13%。lc-ms(esi+)330.9[m+h]+。实施例23抗体(ab)与第一连接子部分(a)偶联物(ab-a)的合成本发明提供的连接子(即第一连接子部分a)在抗体药物偶联物中主要用于与抗体进行偶联,为了验证本发明提供的连接子与抗体的偶联能力,我们将所上述实施例合成的连接子与抗体单独进行偶联,所用的偶联方法为:1)还原剂和保护剂母液配制:用纯化水分别配制还原剂和保护剂如下:1~20mmtcep(tris-2-carboxyethyl-phosphine)(还原剂)、1~20mmdtpa(diethylenetriaminepentacetateacid)(保护剂)母液;2)抗体还原反应:将dtpa和tcep母液分别加入单克隆抗体溶液(如:5~30mg/ml)中。dtpa母液按照一定体积比(1~5:10)混合加入单克隆抗体溶液(如:5~30mg/ml),tcep与单克隆抗体的摩尔比0.5~6.0:1;还原剂与保护剂加入后,于25℃搅拌反应1h;3)第一连接子部分(a)溶液配制:将第一连接子部分(a)溶于25%的dmso(dimethylsulfoxide,二甲亚砜)中,形成浓度为5mm的第一连接子部分(a)溶液;4)第一连接子部分(a)与抗体偶联反应:将步骤3)中的第一连接子部分(a)溶液缓慢加入到步骤(2)中得到的还原后的单克隆抗体溶液中,根据实际需要,控制第一连接子部分(a)与抗体巯基的摩尔比在0.3~5:1区间内,于25℃搅拌反应4h;5)除杂及检测步骤:步骤4)反应结束后,用pbs缓冲液离心超滤3次,纯化去除残余未反应药物以及dmso等游离小分子,并且利用sds-page电泳法检测偶联情况。用sds-page电泳法对抗体药物偶联物的偶联情况进行分析,所用方法如下所示:sds-page检测使用的是nupage4-12%的胶板,取1.5μl样品于1.5mlep管中,加入2.5μlbuffer,再加入1.5μl还原剂,加水定容至10μl,在振荡器上混匀后,置于微波炉上加热1min。上样后开始跑胶,待跑胶结束,取出胶置于保鲜盒中,加入50ml速染染色液,在微波中加热5min后在摇床上染色5min,倒掉染色液后加入100ml纯化水,在电磁炉中加热5min,摇床上脱色1h,重复三次。这里采用的抗体为her2单克隆抗体(抗体序列信息参照中国专利公告号为cn105008398b的专利说明书的第41、42、43段),连接子为:偶联结果如图5所示,图5中,编号1、2、3为her2-a'-7,偶联缓冲液ph分别为8.5、9.0、7.4;编号4、5、6为her2-a'-8,偶联缓冲液ph分别为8.5、7.4、9.0;编号7、8、9为her2-a'-10,偶联缓冲液ph分别为8.5、9.0、7.4;编号10、11、12为her2-a'-11,偶联缓冲液ph分别为9.0、7.4、9.5;编号13、14、15为her2-a'-12,偶联缓冲液ph分别为7.4、9.0、8.5;编号16、17、18为her2-a'-20,偶联缓冲液ph分别为8.5、9.0、7.4;编号19、20、21为her2-a'-28,偶联缓冲液ph分别为9.0、8.5、7.4;编号22、23、24为her2-a'-31,偶联缓冲液ph分别为7.4、8.5、9.0。由于采用的还原电泳,抗体间的巯基全部被打开,因此,图5中的25kd代表抗体的一个轻链的分子量,50kd代表一个重链的分子量,75kd代表一个轻链和一个重链的分子量(即一个连接子与一个重链和一个轻链间的巯基共价连接),100kd代表两重链的分子量(即一个连接子与两个重链间的巯基共价连接),125kd代表一个轻链两个重链的分子量(即一个连接子与两个重链间和一个轻链的巯基共价连接),150kd代表两重链两轻链的分子量(即一个连接子与两个重链和两个轻链间的巯基共价连接)。结果显示,上述抗体药物偶联物均存在一个连接子同时与一个重链和一个轻链、两个重链、两个重链和一个轻链、两个重链和两个轻链间的巯基共价连接的情况。且大部分抗体-连接子偶联物中存在150-75kd的条带,证明抗体内的4对二硫键被完全或部分桥接,且桥接偶联效果较好。从图5中,我们也可以看出,缓冲溶液ph=9.0的条件下,偶联结果较ph=8.5及7.4条件下好。基于本发明提供的连接子与抗体的桥接偶联效果较好,我们进一步选取了a'-7、a'-8、a'-10、a'-11、a'-28进行抗体药物偶联物的制备。所用的抗体为her2单克隆抗体(同上),所用的药物为mmae、mmaf、mmad、cbi、dm1、dm4。对于mmae和mmad而言,本发明涉及的抗体药物偶联物的制备采用一般分为三步:第一步制备第二连接子l与药物d的偶联物l-d;第二步制备第一连接子a与l-d的偶联物a-l-d;第三步制备抗体ab与a-l-d的偶联物,即抗体药物偶联物ab-a-l-d。对于mmaf、cbi、dm1、dm4而言,本发明涉及的抗体药物偶联物的制备采用一般分为两步:第一步制备第一连接子a与药物d的偶联物a-d;第二步制备抗体ab与a-d的偶联物,即抗体药物偶联物ab-a-d。具体的制备方法见如实施例26-29所示。实施例24第二连接子部分(l)与药物(d)组合物(l-d)的合成l-d的合成采用通用方法:将第二连接子部分l溶于适量dmf中,氮气保护下加入适量的药物(d)、hobt、dipea和吡啶。室温搅拌24小时,tlc监测反应进程。反应结束后,用制备型高效色谱进行纯化,制备液经冻干后得到第二连接子部分与药物的偶联物(l-d)。采用上述通用方法制备val-cit-pab-mmae,其合成路线如下所示:取化合物53(即fomc-val-cit-pab-pnp,995.4mg,1.3mmol)和hobt(67.6mg,0.65mmol)于反应瓶中,加入5ml无水dmf溶解,加入dipea(258.5mg,2mmol),常温搅拌10min。将化合物54(即mmae,717mg,1mmol)溶于5ml无水dmf中,滴加反应液中,室温搅拌过夜。反应结束后,过滤,有机相浓缩,残余物经制备液相色谱仪分离得636.2mg化合物55(即fmoc-val-cit-pab-mmae),白色固体,收率47.3%,lc-ms(esi+)1345.8[m+h]+。取化合物55(270mg,0.2mmol),加入20%哌啶的乙腈(v/v)溶液3ml,室温搅拌2.5h,固体逐渐溶解。反应完成后,减压蒸馏除去溶剂,残余物经制备液相色谱仪分离,得214mg化合物62(即val-cit-pab-mmae),收率95%,lc-ms(esi+)1123.7[m+h]+。利用第二连接子部分(l)与药物(d)的合成是合成其他l-d化合物的通用方法,如val-cit-pab-mmad。实施例25第一连接子部分(a)与第二连接子部分-药物组合物(l-d)组合物(a-l-d)的合成a-l-d合成的通用方法:称取适量第一连接子部分、edci、hobt,溶于适量无水dmf中,常温搅拌2h。将适量的自制l-d溶于适量无水dmf中,缓慢滴加至反应液中,室温搅拌4h。反应完成后,反应液用乙腈稀释,制备液相色谱仪分离,洗脱液冻干即得第一连接子部分(a)与第二连接子部分-药物(l-d)的组合物。采用上述通用方法将a'-7与化合物56(val-cit-pab-mmae)进行合成,其合成路线如下所示:将化合物a'-7(27.8mg,0.053mmol)、edci(11.2mg,0.058mmol)、hobt(1.4mg,0.01mmol)溶于0.3ml无水dmf中,常温搅拌2h。将化合物56(即val-cit-pab-mmae,40mg,0.035mmol)溶于0.2ml无水dmf中,滴加至反应液中,室温搅拌4h。反应完成后,反应液用2ml乙腈稀释后,经制备液相色谱仪分离,洗脱液经冻干得8.6mg化合物57(即a'-7-val-cit-pab-mmae),浅黄色固体,收率15%。lc-ms(esi+)1624.6[m+h]+。利用上述通用方法将其他第一连接子部分与l-d化合物进行合成,如a'-8-val-cit-pab-mmae、a'-10-val-cit-pab-mmae、a'-11-val-cit-pab-mmae、a'-11-val-cit-pab-mmad、a'-28-val-cit-pab-mmae等。作为对比,我们也制备了公开号为cn103933575a的中国专利权利要求第7页公开的化合物6(本发明将此化合物定义为575dz)与第二连接子部分(val-cit-pab)和药物部分(mmae)的偶联物575dz-val-cit-pab-mmae。实施例26第一连接子部分(a)与药物(d)组合物(a-d)的合成(1)a'-7-mmaf的合成将化合物a'-7(51.8mg,0.1mmol)、edci(21.1mg,0.11mmol)、hobt(2.7mg,0.01mmol)溶于2ml无水dmf中,常温搅拌2h。将化合物58(即mmaf,48.8mg,0.066mmol)溶于0.8ml无水dmf中,滴加至反应液中,室温搅拌4h。反应完成后,反应液用4ml乙腈稀释后,经制备液相色谱仪分离,洗脱液经冻干得25.9mg化合物a'-7-mmaf,浅黄色固体,收率21%。lc-ms(esi+)1233.4[m+h]+。(2)a'-8-cbi的合成合成方法同a'-7-mmaf的合成方法。加入a'-8(22.0mg,0.047mmol,1.0eq)和化合物59(即cbi,14.0mg,0.020mmol,0.7eq),得到6.0mg白色固体a'-8-cbi,收率21%,lc-ms(esi+)868.0[m+h]+。(3)a'-8-dm1的合成化合物60(45.0mg,0.21mmol,1.0eq)用2ml无水thf溶解,加入1mlsat.nahco3水溶液,滴加入dm1(61,150.0mg,0.21mmol,1.0eq)的1mldmf溶液,室温搅拌2h;加入10ml水稀释,ea萃取,水相用15%柠檬酸溶液调节ph=3,用15mlea萃取,饱和nacl洗涤,干燥,浓缩得165.7mg化合物62,白色固体,收率85%,lc-ms(esi+)949.3[m+h]+。将a'-8(150.0mg,0.33mmol,1.0eq)溶解于5mldmf中,ar气氛下降温至-15℃,加入tea(98.0mg,0.99mmol,3.0eq)的1mldmf溶液,搅拌5min,而后加入t3p(154.0mg,0.48mmol,1.5eq)的1mldmf溶液,搅拌1.5h;将化合物63(46.0mg,0.29mmol,0.9eq)的1mldmf溶液滴加入反应液中,搅拌过夜。反应结束后,加入30mlea,用30%柠檬酸调节ph=3,萃取,洗涤,干燥,浓缩,经正向柱层析纯化得90mg化合物64,白色固体,收率52%,lc-ms(esi+)607.1[m+h]+。化合物64(90mg,0.15mmol,1.0eq),加入3ml无水dcm搅拌溶解,再加入hcl的1,4-二氧六环溶液(4m,1.0ml,4mmol,26.7eq),室温搅拌2h;减压浓缩,用20ml甲基叔丁基醚超声分散,离心,弃去清液,再加入20ml石油醚分散,离心,弃去清液。固体经减压蒸馏除去溶剂,得82mg化合物65,白色固体,收率100%,lc-ms(esi+)506.9[m+h]+。将化合物62(30.0mg,0.03mmol,1.0eq)溶解于2mldmf中,ar气氛下降温至-15℃,加入tea(11.0mg,0.095mmol,4.0eq)的500μldmf溶液,搅拌5min,而后加入t3p(15.0mg,0.047mmol,1.5eq)的500μldmf溶液,搅拌1.5h;将化合物65(19.0mg,0.035mmol,1.1eq)的500μldmf溶液滴加入反应液中,搅拌过夜。反应结束后,反应液用2ml乙腈稀释,经制备液相纯化,得9mg产品a'-8-dm1,收率20%。lc-ms(esi+)1437.3[m+h]+。(4)a'-7-dm4的合成将67(737.5mg,4.03mmol)、edci(846.13mg,4.43mmol)、hobt(108.9mg,0.806mmol)溶于10mldcm中,室温搅拌2h;将66(1g,4.03mmol)溶于5ml二氯甲烷中,分3次缓慢滴加至反应体系中,室温搅拌过夜。反应结束后,减压浓缩除去溶解,残余物用6ml乙腈/水(1:1)溶解,制备分离,冻干得化合物681.11g,无色油状物,收率67%。lc-ms(esi+)414.2[m+h]+。将化合物68(1.11g,2.7mmol)用1.1mldma溶解备用,取0.1ml溶于1mldma中,加入dipea(41.3mg,0.32mmol)室温搅拌2min,加入dm4(69,202.5mg,0.26mmol),室温搅拌6h,反应液直接制备分离,冻干后得260mg化合物70,白色固体,收率84%。lc-ms(esi+)1193.5[m+h]+。化合物71的合成方法同化合物65。加入119.3mg化合物70,得106mg化合物71,白色固体,收率96%。lc-ms(esi+)1093.5[m+h]+。a'-7-dm4的合成方法同a'-8-dm1。加入52mga'-7和54.6mg化合物71,得15mga'-8-dm1,白色固体,收率19%。lc-ms(esi+)1594.4[m+h]+。实施例27抗体药物偶联物合成及产物均一性分析抗体药物偶联物合成的通用方法为:1)还原剂和保护剂母液配制:用纯化水分别配制还原剂和保护剂如下:1~20mmtcep(tris-2-carboxyethyl-phosphine)(还原剂)、1~20mmdtpa(diethylenetriaminepentacetateacid)(保护剂)母液;2)抗体还原反应:将dtpa和tcep母液分别加入单克隆抗体溶液(如:5~30mg/ml)中。dtpa母液按照一定体积比(1~5:10)混合加入单克隆抗体溶液(如:5~30mg/ml),tcep与单克隆抗体的摩尔比0.5~6.0:1;还原剂与保护剂加入后,于25℃搅拌反应1h。3)a-l-d合成及溶液配制:合成第一连接子部分-第二连接子部分-药物部分偶联物(a-l-d),将合成的a-l-d溶于25%的dmso(dimethylsulfoxide,二甲亚砜)中形成浓度为5mm的a-l-d溶液;4)药物与抗体偶联反应:将步骤3)中的a-l-d溶液缓慢加入到步骤(1)中得到的还原后的单克隆抗体溶液中,根据实际需要,控制a-l-d与抗体巯基的摩尔比在0.3~5:1区间内,于25℃搅拌反应4h。5)除杂及检测步骤:步骤4)反应结束后,用pbs缓冲液离心超滤3次,纯化去除残余未反应药物以及dmso等游离小分子,并且利用sds-page电泳和疏水高效液相(hic-hplc)法检测偶联情况。用sds-page电泳和hic-hplc法对抗体药物偶联物的偶联情况进行分析,所用方法如下所示:1)聚丙烯酰胺凝胶电泳(sds-page)sds-page检测使用的是nupage4-12%的胶板,取1.5μl样品于1.5mlep管中,加入2.5μlbuffer,再加入1.5μl还原剂,加水定容至10μl,在振荡器上混匀后,置于微波炉上加热1min。上样后开始跑胶,待跑胶结束,取出胶置于保鲜盒中,加入50ml速染染色液,在微波中加热5min后在摇床上染色5min,倒掉染色液后加入100ml纯化水,在电磁炉中加热5min,摇床上脱色1h,重复三次。2)疏水作用色谱(hic)分析测试条件:hic-hplc:东曹tsk凝胶丁基,4.6mm×3.5厘米,10毫米;缓冲液a:20mm磷酸钠,1.5m硫酸铵,ph值为7.0;缓冲液b:20mm磷酸钠,25%(v/v)异丙醇(isopronal),ph为7.0;流速:1毫升/分钟;梯度:15分钟内10%缓冲液b至100%缓冲液b;10μl样品。我们利用上述方法制备了如下5种抗体药物偶联物,它们分别是:her2-a'-7-val-cit-pab-mmae、her2-a'-11-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae、her2-575dz-val-cit-pab-mmae,其中抗体为her2单克隆抗体(同上),用hic-hplc法和sds-page电泳对上述4种抗体药物偶联物的偶联情况进行分析,结果如表1、图1a、图1b、图1c、图1d和图2a、图2b所示。表1和图1a、图1b、图1c、图1d中,d0%代表一个抗体通过连接子偶联上0个药物的抗体药物偶联物的比例,即裸抗的比例,d1%代表一个抗体通过连接子偶联上1个药物的抗体药物偶联物的比例,d2%代表一个抗体通过连接子偶联上2个药物的抗体药物偶联物的比例,d3%代表一个抗体通过连接子偶联上3个药物的抗体药物偶联物的比例,d4%代表一个抗体通过连接子偶联上4个药物的抗体药物偶联物的比例,d5%代表一个抗体通过连接子偶联上5个药物的抗体药物偶联物的比例。结果显示,her2-a'-7-val-cit-pab-mmae的平均dar值为4.10(偶联时药物与抗体的摩尔比为5:1);her2-a'-11-val-cit-pab-mmae的平均dar值为2.12(偶联时药物与抗体的摩尔比为5:1);her2-a'-10-val-cit-pab-mmae的平均dar值为2.26(偶联时药物与抗体的摩尔比为5:1);her2-575dz-val-cit-pab-mmae的平均dar值为4.10(偶联时药物与抗体的摩尔比为5:1)。结果显示,her2-a'-7-val-cit-pab-mmae、her2-a'-11-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae均表现出了良好的偶联效果(理想状态下平均dar值为2-4之间),其中her2-a'-11-val-cit-pab-mmae和her2-a'-10-val-cit-pab-mmae的dar2比例分别为65.574%和78.9%,her2-a'-7-val-cit-pab-mmae的d4%比例为59.45%,dar值分布非常集中,产品均一性非常好(均优于her2-575dz-val-cit-pab-mmae)。表1抗体药物偶联物疏水作用色谱(hic-hplc)分析结果adcd0%d1%d2%d3%d4%d5%darher2-a'-7-val-cit-pab-mmae00.071.9312.1659.4526.404.10her2-a'-10-val-cit-pab-mmae09.0378.911.9002.26her2-a'-11-val-cit-pab-mmae011.4165.5723.02002.12her2-575dz-val-cit-pab-mmae001.9918.4652.9421.174.10图2a、图2b给出了上述抗体药物偶联物的sds-page电泳图谱。其中图2a给出了her2-a'-11-val-cit-pab-mmae(偶联缓冲液ph分别为8.5、9.0、7.4)、her2-a'-7-val-cit-pab-mmae(偶联缓冲液ph分别为8.5、9.0、7.4)、her2-a'-10-val-cit-pab-mmae(偶联缓冲液ph分别为8.5、9.0、7.4)的sds-page电泳图谱,图2b从左到右依次为her2-a'-7-val-cit-pab-mmae、her2-575dz-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae的sds-page电泳图谱(偶联缓冲液ph为9.0)。由于采用的还原电泳,抗体间的巯基全部被打开,因此,25kd代表抗体的一个轻链的分子量,50kd代表一个重链的分子量,75kd代表一个轻链和一个重链的分子量(即一个连接子与一个重链和一个轻链间的巯基共价连接),100kd代表两重链的分子量(即一个连接子与两个重链间的巯基共价连接),125kd代表一个轻链两个重链的分子量(即一个连接子与两个重链间和一个轻链的巯基共价连接),150kd代表两重链两轻链的分子量(即一个连接子与两个重链和两个轻链间的巯基共价连接)。图2a显示出,本实施例提供的抗体药物偶联物(her2-a'-7-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae、her2-a'-11-val-cit-pab-mmae)均存在一个连接子同时与一个重链和一个轻链(75kd)、两个重链(100kd)、两个重链和一个轻链(125dd)、两个重链和两个轻链(150kd)间的巯基共价连接的情况,证明抗体内的4对二硫键被完全或部分桥接,且桥接偶联效果较好。图2b给出了her2-a'-7-val-cit-pab-mmae、her2-575dz-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae的平行对比sds-page电泳图谱(偶联缓冲液ph为9.0),从图中,我们可以看出,her2-a'-7-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae偶联物中150kd和125kd条带比例明显高于her2-575dz-val-cit-pab-mmae相应条带;同时,50kd和25kd条带的比例也明显降低,证明本发明中的第一连接子片段具有较好的偶联效率,能够得到桥接比例更高的抗体-药物偶联物。实施例28抗体药物偶联物的水解分析我们采用了分子量分析和蛋白等电点分析对抗体药物偶联物(claudin18.2-a’-7-vc-pab-mmae)的水解情况进行表征。1)分子量分析(lc-ms)测试条件:仪器型号:thermoqexactiveplus;流动相:25mm乙酸铵溶液;流速:0.3ml/min;进样量:1mg/ml,20μl,即20μg;检测范围:5000-8000da;输出范围:140000-160000da。2)等电点分布分析仪器信息:成像毛细管等电聚焦电泳仪,型号:ice3。仪器设置:聚焦时间1:1500vfor1.00min;聚焦时间2:3000vfor8.00min。裸抗体(claudin18.2抗体):两性电解质载体:1%pharmalyte5-8,3%pharmalyte8-10.5;低等电点标记物:8.18;高等电点标记物:9.77;蛋白浓度:0.25mg/ml;托盘温度:15℃。claudin18.2-a’-7-vc-pab-mmae:两性电解质载体:3%servalyt5-9,1%servalyt9-11;低等电点标记物:5.85;高等电点标记物:9.46;蛋白浓度:0.50mg/ml;托盘温度:15℃。图6a给出裸抗体的分子量结果,图6b给出claudin18.2-a’-7-vc-pab-mmae的分子量结果。从图中我们可以看出,裸抗体的分子量为145144da,claudin18.2-a’-7-vc-pab-mmae的分子量(a’-7-vc-pab-mmae与抗体的比例为4:1时)为152078da、152119da、152147da。当一个抗体偶联4个a’-7-vc-pab-mmae且未发生水解的情况下,其理论分子量为151999da。实际检测到的分子量包括152078da、152119da、152147da,与理论分子量有较大偏差,超出仪器偏差范围(20da)。当claudin18.2-a’-7-vc-pab-mmae偶联物发生水解且水解4个、7个、8个马来酰亚胺时,其理论分子量为152071da、152125da、152143da,与实测值相比,相对偏差在仪器的偏差范围内,推测抗体-a’-7-vc-pab-mmae发生部分水解。接下来,我们又对水解部分进行了研究。图7a给出裸抗体的等电点分布结果,图7b给出claudin18.2-a’-7-vc-pab-mmae的等电点分布结果。从图7a中我们可以看出,裸抗体的等电点主峰为8.91,其酸性峰(等电点<8.91)和碱性峰(等电点>8.91)比例相对较低。从图7b中我们可以看出,抗体-a’-7-vc-pab-mmae的等电点分布在7.86-8.70之间。相对于裸抗体,抗体-a’-7-vc-pab-mmae的等电点明显下降,说明其表面分布有更多的酸性基团,例如羧基,结合结构分析,发生水解的位置在抗体-a’-7-vc-pab-mmae中的马来酰亚胺部分,并且通过控制抗体药物偶联物的合成水解环境可实现对抗体药物偶联物的连接子中马来酰亚胺的水解程度的控制,如实现完全水解或不水解。此外,也可以结合纯化手段,从合成产物中分离实现相同水解程度的产物。实施例29抗体药物偶联物对抗原的亲和力测定(elisa法)通用步骤:用包被缓冲液稀释抗原至需要的浓度(如500ng/ml、250ng/ml、100ng/ml,也可根据实验需要自行安排)100μl/孔,4℃过夜;洗板:pbst洗涤液350μl/孔洗板3次,拍干;封闭:用3%bsa封闭液200μl/well封闭,4℃过夜;洗板:pbst洗涤2遍,300μl/well,吸干孔内溶液;加样:用稀释液1%bsa-pbst溶液将样品稀释,将稀释好的样品以100μl/well加入封闭后的细胞培养板中,设置三个复孔,样品稀释液作为空白对照,37℃培养箱孵育2小时(稀释方案可根据实际情况进行改变);洗板:pbst洗涤2遍,200μl/well,吸干孔内溶液;加入检测抗体:用1%bsa-pbst以1:5000稀释goatanta-humanigg-fchrpconjugated,100μl/孔,37℃育1小时(稀释液可根据实际情况进行改变);洗板:pbst洗涤4遍,200μl/well,吸干孔内溶液;显色:100μl/well加入tmbsubstrate,显色2分钟;终止:50μl/well加入2mh2so4,终止反应;读数:酶标仪测定450/655nm处光密度吸收值,实验结果用prism分析软件进行分析,以浓度为横坐标,光密度吸收值为纵坐标,由软件自动计算ec50等值,结果见表2和图3。表2表示的是抗体药物偶联物(her2-a'-7-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae)对her2抗原的ec50值。其中her2为裸抗体,her2-mc-vc-pab-mmae为对照组adc。结果显示,her2-a'-10-val-cit-pab-mmae和her2-a'-7-val-cit-pab-mmae对抗原的亲和力均较好,数据结果显示所采用的连接方式未改变抗体原来的亲和力。表3抗体药物偶联物对抗原的亲和力(ec50值)adcec50(ng/ml)r2her21.0730.999her2-mc-vc-pab-mmae2.1320.994her2-a'-10-val-cit-pab-mmae1.7250.999her2-a'-7-val-cit-pab-mmae2.6921.000实施例30体外药效实验将装有人乳腺癌sk-br-3细胞和培养基的培养皿,置于37℃、5%co2培养箱中培养,取生长状态良好的细胞,弃去原培养基,分别通过培养基重悬并计数;将细胞悬液加入到96孔板中(每孔细胞数为5000个),37℃、5%co2培养箱孵育过夜;药物稀释并加药培养,培养72h后采用cellcountingkit-8(简称cck-8试剂盒)进行活性显色,显色后的96孔板通过酶标仪检测450nm处od值。prism软件通过od值计算得出ic50值,软件以抑制率作为y值,药物浓度作为x值进行四参数曲线拟合,并记录最大抑制率与最小抑制率中间的抑制率值所对应的药物浓度值(软件默认为ic50值)。当拟合的曲线呈“s型曲线”且r2≥0.95时,ic50值有效,抗体药物偶联物her2-a'-8-val-cit-pab-mmae、her2-a'-7-val-cit-pab-mmae、her2-a'-11-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae对人乳腺癌sk-br-3细胞的ic50值见表3所示,其抑制率曲线图如图4a和图4b所示。表3表示的是抗体药物偶联物(her2-a'-8-val-cit-pab-mmae、her2-a'-7-val-cit-pab-mmae、her2-a'-11-val-cit-pab-mmae、her2-a'-10-val-cit-pab-mmae)对人乳腺癌sk-br-3细胞的ic50值抑制率。表3抗体药物偶联物对人乳腺癌sk-br-3细胞体外药效本发明已通过各个具体实施例作了举例说明。但是,本领域普通技术人员能够理解,本发明并不限于各个具体实施方式,普通技术人员在本发明的范围内可以作出各种改动或变型,并且在本说明书中各处提及的各个技术特征可以相互组合,而仍不背离本发明的精神和范围。这样的改动和变型均在本发明的范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1