结合前列腺特异性膜抗原的嵌合抗原受体的制作方法
结合前列腺特异性膜抗原的嵌合抗原受体
[0001]
本发明涉及结合肿瘤抗原的嵌合抗原受体,其中该抗原是前列腺特异性膜抗原(psma)。将嵌合抗原受体(以下为car)带入免疫细胞,特别是t细胞,nk细胞,inkt细胞和cik细胞中,然后它们与表达psma的肿瘤细胞特异性反应,从而导致肿瘤细胞的消除。本发明的构建体含有两个主要部分。在一方面是特异性结合前列腺特异性膜抗原(psma)的抗原结合区,而另一方面是源自免疫细胞受体负责信号转导和激活免疫细胞的共刺激域。
[0002]
前列腺癌仍然是全世界男性中第二常被诊断出的癌症,每年估计有110万新病例。此外,预计有307,000例死亡,它代表癌症死亡的第五大原因。尽管原发性肿瘤可以成功治疗,但尚无晚期阶段的治愈性治疗。因此,迫切需要新的治疗选择。
[0003]
前列腺特异性膜抗原(psma)是前列腺癌中用于基于抗体的诊断和治疗干预的最佳表征的抗原。该蛋白质也称为谷氨酸羧肽酶ii(ec 3.4.17.21),n-乙酰基连接的酸性二肽酶i(naaladase)或叶酸水解酶。psma是一种ii型膜糖蛋白,其由750个氨基酸(aa)组成,具有19aa的小胞内域,24aa的跨膜域和707aa的大胞外域。胞外域折叠成三个不同的域:蛋白酶域(aa 57
–
116和352
–
590),顶端域(aa 117
–
351)和c端域(aa 591
–
750)。它显示出与人转铁蛋白受体1的高度结构相似性和同一性。psma高度局限于前列腺癌细胞的表面,在所有肿瘤阶段期间存在于癌细胞上,并且在雄激素非依赖性和转移性疾病中表达增强。psma不分泌到胞外空间中,并且经历组成型内在化,这通过psma特异性抗体的结合而增强。这些特征使其成为晚期前列腺癌的靶向治疗的理想候选者。此外,还发现psma实际上在所有实体瘤类型的新生血管内皮中表达而在正常血管内皮中不表达。因此,它被认为是独特的抗血管生成靶标。
[0004]
单克隆抗体(mab)是用于细胞靶向的高度特异性且用途广泛的工具。在过去的几十年中,它们在医学研究中引起了高度兴趣,并已成为发展最快的一类治疗多种人类疾病的药物,所述疾病包括癌症。抗体7e11是第一个发布的psma特异性mab,并且被发现与psma胞内域的n端(mwnllh)结合。7e11的标签内(in-labeled)形式(prostascint,cytogen,philadelphia,pa)已获得美国食品药品管理局(fda)的批准,用于软组织中转移性前列腺癌的检测和成像。但是,由于抗体结合胞内表位,因此7e11无法结合活细胞。使用prostascint进行体内成像的阳性信号只得追溯到检测到肿瘤块内死亡或垂死的细胞。因此,产生了一类新的抗-psma mab,其特异性结合活细胞表达的psma的胞外表位。
[0005]
ep 1 883 698公开了三种不同的mab 3/a12、3/e7、3/f11,它们显示出与前列腺癌细胞和前列腺组织标本表面上的psma的细胞外部分的强特异性结合。与经过临床验证的放射免疫疗法抗体mab j591(pmid:18552139;pmid:24135437;pmid:25771365;pmid:26175541)直接比较,mab 3/f11与表达psma的c4-2前列腺癌靶细胞具有更高的结合力(3/f11的k
d
[定义为平均半最大饱和浓度]=9nm;j591的k
d
=16nm)。此外,竞争性结合研究表明,mab 3/f11与j591(pmid:19938014)结合不同的胞外psma表位。在一组人体正常组织的免疫组织学研究中,检测不到3/f11 mab与psma阴性组织(肾上腺、骨髓、小脑、大脑、垂体、结肠、食道、心脏、肾脏、肝脏、肺、心包的间皮细胞、神经、卵巢、胰腺、骨骼肌、皮肤、脾、胃、睾丸、胸腺、甲状腺、扁桃体和子宫)的结合。仅观察到与唾液腺分泌细胞和十二指肠刷状缘
细胞的结合,已知它们表达psma(pmid:19938014)。mab 3/f11对所有测试的正常前列腺组织的腺泡分泌上皮细胞显示中等免疫反应性。几乎在腺癌的所有上皮细胞以及淋巴结转移中都观察到了更强烈且广泛的染色。在乳腺标本的冷冻切片上未检测到免疫组织学染色。相反,根据其他已发表的数据,用mab j591检测到乳腺导管上皮的染色。由于通过pcr或者通过western印迹法都检测不到乳腺组织中的psma表达,因此mab j591可能与另一种抗原发生交叉反应。
[0006]
通过噬菌体展示技术从mab 3/f11产生单链可变片段(scfv)d7(如ep 1 883 698 b1中所公开)。对于抗psma scfv的特异性最重要的片段是v
l
和v
h
部分,它们优选用聚甘氨酸接头相连。d7以约18nm的k
d
与表达psma的c4-2细胞结合,并且与亲本mab 3/f11的预温育完全抑制结合活性。这证明scfv d7与3/f11结合相同的psma表位。scfv d7及其人源化形式已成功用于构建基于假单胞菌外毒素a(pe)的免疫毒素,该免疫毒素对表达psma的前列腺癌细胞表现出高且特异性的细胞毒性,并且在携带前列腺肿瘤的小鼠中显示体内抗肿瘤活性。
[0007]
根据本发明,d7 scfv用于构建嵌合抗原受体(car)以提供用于免疫细胞中以靶向表达psma的癌细胞的构建体。
[0008]
ma等人[the prostate(2014)74,pp 286-296]公开了针对psma的第二代car,其包含cd28共刺激域和cd3ζ信号传导域以及来自小鼠抗人psma单克隆抗体3d8的抗原结合部分,所述单克隆抗体3d8可购自northwest biopharmaceutics,inc。
[0009]
santoro等人[cancer immunol.res.(2015),pp 68-84]描述了带有针对前列腺特异性膜抗原的嵌合抗原受体的t细胞,其中psma结合部分源自抗体j591。
[0010]
zhong等人[molecular therapy(2010),pp.413-420]还公开了嵌合抗原受体,其中psma结合片段也源自抗体j591。j591的序列是本领域公知的。wo 2009/017823公开了其vh和vl域。
[0011]
提供了抗原结合片段d7的氨基酸序列(图10;seq id no:1)和核酸序列(seq id no:8),包括编码所述构建体的互补链(seq id no:9)。此外,显示并用灰色箭头突出显示抗原结合片段的高度重要的部分,即cdr:(cdr-h1(seq id no:2),cdr-h2(seq id no:3),cdr-h3(seq id no:4),cdr-l1(seq id no:5),cdr-l2(seq id no:6)和cdr-l3(seq id no:7))。
[0012]
鼠抗体的人源化涉及将有益特性(例如抗原特异性结合,通过与其他抗原的非交叉反应性避免脱靶效应)从一种抗体转移到另一种抗体以降低免疫原性。人源化通常是人类使用所必需的,因为患者通常以针对非人类抗体的免疫反应响应,这可以导致治疗无效,并且最坏的情况是危及生命。人源化的构建体可源自图10所示的抗原结合片段的序列。cdr区在结构上限定互补位,即抗原结合片段与抗原的接触位点。序列的剩余部分编码框架区,其形成互补位的支架。对于人源化过程(例如通过计算机建模),首先将框架序列与源自人的其他抗原结合序列进行比较。通常,选择与图10所示框架序列具有最高相似性的人序列(受体框架)。将cdr区接枝到人受体框架中,以消除可能导致不期望的人抗小鼠抗体(hama)免疫反应的氨基酸序列。针对前瞻性回复突变分析潜在关键位置上的取代(例如负责折叠互补位或vh-vl界面的氨基酸)。即使在cdr序列中,也可以对氨基酸进行特殊的修饰,以避免免疫原性,确保互补位的正确折叠,并维持抗原特异性结合。
[0013]
在抗体的人源化过程中,优选地在与相应鼠序列具有最高同源性的人免疫序列中选择序列。然后确定cdr的位置。cdr的确定在本领域中是公知的,并且应当注意的是,用于确定的不同方法是已知的,由此cdr的位置可能有所不同。在本发明的过程中,使用根据kabat的cdr确定以及根据imgt(international immunogene ticks)的确定。
[0014]
在本发明的一个优选实施方案中,人源化是根据所谓的“cdr接枝”进行的。优选地,根据imgt方法确定功能性cdr,并且将那些cdr转移到与起始鼠抗体具有最高序列同源性的人框架区中。然后,关于诸如大小,极性或电荷的生化特性确定关于人源化和鼠抗体的框架区中的单一氨基酸的差异。在第一步中,调整相似的氨基酸,并随后改变不同的氨基酸,从而以完整人框架结束。
[0015]
由于本文公开的人源化形式已经完全或在很大程度上保留cdr,因此人源化变体具有鼠抗体的相同功能,但是,亲和力可能彼此略有不同。
[0016]
图11中显示了人源化实验的结果。结果证明,应维持cdr-h1,cdr-h3和cdr-l3而不进行任何修改。在cdr-l2中,可以替换一个氨基酸,其指示为x9。x9可以具有脂肪族、不带电荷的氨基酸的含义,其可以是甘氨酸、丙氨酸、缬氨酸、赖氨酸、异赖氨酸和/或脯氨酸。
[0017]
在cdr-l1中,可以替换两个氨基酸,其称为x7和x8。那些氨基酸可以是亲水性、不带电荷的氨基酸,例如丝氨酸、苏氨酸、天冬酰胺和/或谷氨酰胺。
[0018]
最高的柔性似乎具有cdr-h2,其中可以替换多达六个氨基酸。那些氨基酸称为x1、x2、x3、x4、x5和x6。用于人源化抗体或抗原结合片段中的替换的氨基酸具有以下含义:
[0019]
x1、x4、x6:亲水的不带电荷的氨基酸[丝氨酸(s)、苏氨酸(t)、天冬酰胺(n)、谷氨酰胺(q)]
[0020]
x2,x3:脂肪族的不带电荷的氨基酸[甘氨酸(g)、丙氨酸(a)、缬氨酸(v)、亮氨酸(l)、异亮氨酸(i)、脯氨酸(p)]
[0021]
x5:碱性氨基酸[组氨酸(h)、赖氨酸(k)、精氨酸(r)]。
[0022]
近年来,已引入过继免疫细胞疗法作为一种新颖的构思,通过重定向免疫系统消除肿瘤细胞来治疗不同的癌症。最成功的构思之一是基于t细胞的遗传工程来表达以人类白细胞抗原(hla)非依赖性的方式结合肿瘤抗原或肿瘤相关抗原的嵌合抗原受体(car)。靶向cd19的car t细胞已成功用于治疗b细胞急性成淋巴细胞性白血病(b-all),在几项临床试验中,>90%的患者已完全消退。基于此成功,已经开展了超过200次临床试验来主要治疗血液系统恶性疾病。然而,对于实体瘤,迄今为止,car t细胞疗法的效力似乎相当低。此失败的主要原因似乎是肿瘤微环境(tme),它是肿瘤存在的细胞环境。它包括各种免疫细胞、成纤维细胞、细胞外基质(ecm)以及周围的血管。已经描述了许多描述tme中细胞毒性t细胞活性的限制的机制,包括激活基于pd-1的t细胞免疫检查点抑制。结合t细胞检查点拮抗剂克服这些限制有助于改善tme中的抗肿瘤活性。
[0023]
如本发明所用,根除肿瘤需要肿瘤抗原特异性免疫细胞,优选t细胞的充分存活和肿瘤内激活。为了满足这些要求,在抗原引发和刺激时必须给t细胞适当的激活信号。因此,本发明的嵌合抗原受体组合抗原结合片段作为t细胞上受体的一部分。抗原结合片段结合t细胞应结合的特异性抗原(此处为psma)。此外,这些受体分别含有来自cd28和4-1bb的序列作为共刺激信号传导域。已经显示了向基于cd3ζ链的受体添加cd28序列或其他共刺激性信号传导域增加抗原诱导的白介素2分泌和体外t细胞扩充。在当前情况下,信号传导域由cd3
ζ域和胞内cd28或4-1bb域组成。
[0024]
通常,car的设计可以存在变化,同时已知几代car。car系统的主要成分是t细胞受体(tcr)复合物的cd3ζ胞内域、跨膜域、铰链区和抗原结合部分。在car的设计中,抗原结合域连接至铰链区(该铰链区也称为间隔区)、跨膜域和胞质域。那些部分负责抗原结合部分的位置,在t细胞膜上的附着以及胞内信号传导。除了car设计中的这种结构规则外,铰链区的形态特征(如其长度和序列)对于有效靶向也是重要的。胞内域作用为信号转导物。由于激活的t细胞和静止的t细胞的功能不同,cd3ζ的胞质区段起主要作用。然而,此胞质部分单独不能激活静止的t细胞。因此,需要至少一个次级信号用于t细胞的完全激活。在本发明中,优选使用4-1bb或cd28共刺激域。或者,可以使用其他共刺激域,例如源自例如cd27、icos和ox40的共刺激域。
[0025]
在优选的实施方案中,将突变引入人igg1 fc铰链区中,从而避免了诸如防止lck激活或固有免疫应答的意外启动的副作用。那些突变之一避免lck结合,而另一个突变可以抑制treg细胞与构建体的结合。此类突变可以改善构建体的生物学活性。
[0026]
为了治疗人类患者,必须从个体患者的外周血中富集t细胞(自体环境)或由供体提供(同种异体环境)。这可以例如通过白细胞单采术来完成。然后用包含car的遗传信息的合适载体离体转染或转导富集的t细胞。
[0027]
将编码car的遗传信息插入合适的载体中。此类载体优选是慢病毒或逆转录病毒载体。目前认为转导原代t细胞的金标准是慢病毒载体,它似乎是较简单的逆转录病毒载体的有效替代品。作为慢病毒载体的另一种选择,可以在转座子或质粒的帮助下将信息引入t细胞。病毒和非病毒递送两者的替代方法是最近描述的基因编辑工具,称为crispr/cas或其他设计者核酸酶,例如转录激活物样效应核酸酶(talen)或锌指核酸酶(zfn)。该技术平台提供了以靶向方式靶向几乎所有基因组位点的可能性。在crispr/cas的情况下,编辑复合物包含cas核酸酶和指导rna,通常由crispr rna(crrna)和反式作用crrna组成。指导rna与靶序列杂交后,cas9(或另一种cas核酸酶,诸如例如cpf1/cas12a)产生双链断裂,其可通过非同源末端连接(nhej)(可以导致基因组基因座功能丧失的事件)进行修复。在合适的供体dna存在下,通过同源性指导修复(hdr)的机制,可以将外源序列(car序列)引入靶定基因座中。这可以用于在不干扰内源基因功能的期望基因组基因座中递送car表达盒,从而最小化用整合病毒载体经历的遗传毒性效应。在另一个优选的实施方案中,使用基因组编辑将car编码序列置于内源启动子的控制下。来自内源启动子(例如trac基因座的启动子)的表达可确保car构建体的最佳表达水平以实现其功能。编码本发明car的核酸序列优选针对人密码子使用进行优化。特别优选的实施方案是编码car28构建体的seq id no:10和编码car41构建体的seq id no:11。
[0028]
在本发明的另一个优选的实施方案中,将编码car构建体的rna引入靶细胞,例如t细胞中。可以通过物理程序,例如电穿孔或通过将细胞膜与合适的囊泡融合,将编码car构建体的核酸引入靶细胞。在该实施方案中,car构建体优选在t细胞中瞬时表达。优点在于,然后存在有经转导的t细胞群体,其仅在短暂的时间内存在于治疗的患者中。
[0029]
在其他优选的实施方案中,将car构建体引入自然杀伤细胞(nk),不变的自然杀伤t细胞(inkt),多样自然杀伤细胞(diverse natural killer cells,dnkt),细胞因子诱导的杀伤细胞(cik)或γ-δt细胞中。此外,可以使用合适的同种异体细胞。
[0030]
在另一个优选的实施方案中,除了本文所述的car构建体之外,还可将调节免疫系统的另一种转基因,例如编码细胞因子,趋化因子受体和/或检查点抑制剂的基因引入免疫细胞中。在另一个优选的实施方案中,基因组编辑用于破坏调节免疫系统的基因的表达,例如编码细胞因子、趋化因子受体和/或检查点抑制剂的基因。
[0031]
在另一个实施方案中,根据本发明的构建体和包含此类构建体的免疫细胞可以用于具有靶向肿瘤注射的局部疗法。在该实施方案中,其优选用将car t细胞施加到患者体内局部肿瘤区域定位的某些位置的自动装置来进行。然后用活检针取出样品,从而形成小腔。在该腔中引入经转导或转染的t细胞,然后抽出针。当存在用常规方法极其难以治疗的实体瘤时,该实施方案是特别有利的。
[0032]
本文公开的嵌合抗原受体可用于治疗与psma表达有关的疾病。psma在源自前列腺癌的肿瘤细胞中表达。存在有已知的前列腺癌的几个阶段,但似乎psma是最适合治疗前列腺癌的标志物之一。术语“前列腺癌”包括源自原发性肿瘤或转移性肿瘤或循环的肿瘤细胞的所有形式的前列腺癌细胞。在一个特别优选的实施方案中,用本发明的嵌合抗原受体工程化改造的免疫细胞用于抵抗表达psma的实体瘤的新血管形成。
[0033]
在本发明的另一个实施方案中,用本文公开的嵌合抗原受体工程化改造的免疫细胞与治疗剂,特别是细胞毒剂组合使用。用于治疗前列腺癌的细胞毒剂是已知的。优选地,此类物质包括紫杉醇(taxol)衍生物、5-氟尿嘧啶、环磷酰胺、米托蒽醌(mitoxantrone)、多西他赛(docetaxel)、卡巴他赛(cabazitaxel)和依托泊苷(etoposide)。下列药物已被批准用于前列腺癌,并优选使用:醋酸阿比特龙(abiraterone acetate)、阿帕鲁胺(apalutamide)、比卡鲁胺(bicalutamide)、卡巴他赛、casodex(比卡鲁胺(bicalutamide))、地加瑞克(degarelix)、多西他赛、eligard(醋酸亮丙瑞林)、恩杂鲁胺(enzalutamide)、erleada(阿帕鲁胺(apalutamide))、firmagon(degarelix)、氟他胺(flutamide)、醋酸戈舍瑞林、jevtana(卡巴他赛)、醋酸亮丙瑞林、lupron depot(醋酸亮丙瑞林)、盐酸米托蒽醌、nilandron(尼鲁米特)、尼鲁米特、provenge(sipuleucel-t)、二氯化镭223、sipuleucel-t、泰素帝(taxotere)(多西他赛)、xofigo(二氯化镭223)、xtandi(恩杂鲁胺)、zoladex(醋酸戈舍瑞林)、zytiga(醋酸阿比特龙)。应当理解,本发明的嵌合抗原受体也可以与用于治疗激素敏感性形式的前列腺癌的药物例如醋酸亮丙瑞林组合使用。
[0034]
在本申请的附图和实施例中进一步描述和说明了本发明的优选实施方案。除非明确排除,否则附图或实施例中公开的所有方面均与本发明有关。在实验部分中公开的本发明的单个特征可以被组合,除非有技术原因反对此类组合。具体而言,图显示了以下实验结果:
[0035]
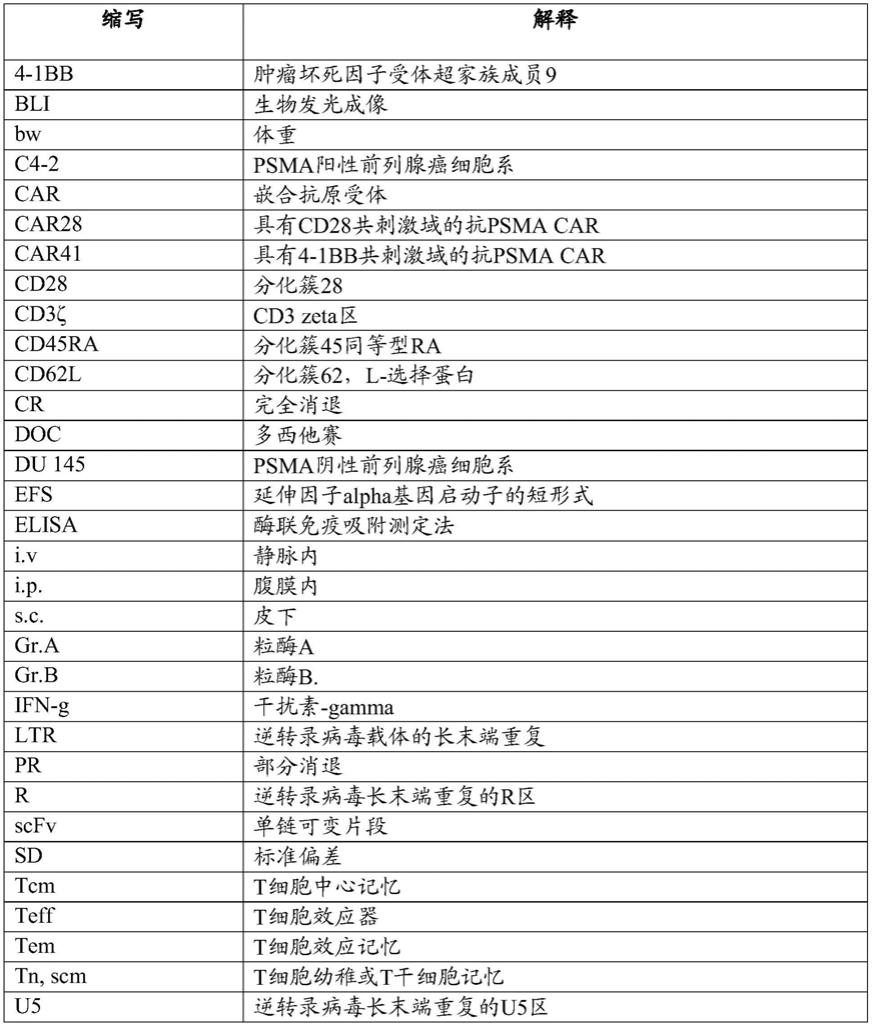
在附图和实验中,使用了以下缩写:
[0036][0037][0038]
图1.psma靶向car t细胞的产生和质量评估。
[0039]
(a)表达第二代抗psma car的自我失活gamma-逆转录病毒载体的示意图。car表达由efs启动子驱动。car由单链可变片段d7(源自针对psma的3f/11鼠单克隆抗体)、铰链区(未显示)、跨膜域、源自4-1bb(car41)或cd28(car28)的共刺激域、以及源自cd3ζ链的胞内信号传导域组成。这两种car中都包含的铰链区源自人igg,并且在d7-scfv和跨膜域之间提供物理间隔区,以实现最佳靶标识别。
[0040]
(b)car表达。在逆转录病毒转导之前用抗cd2/3/28抗体将t细胞激活2-3天。病毒转导后,将t细胞扩充8-9天,然后收获细胞并用抗人igg抗体(car)和cd3染色以评估转导效率。
[0041]
(c)用流式细胞仪分析的定性car t细胞表型表征。通过评估cd62l和cd45ra表达评估t细胞亚组,对于未转导的t细胞(ut)(未显示)在car-cd3+上或者对于这两类car t细胞(未显示)在car+cd3+上预先门控。
[0042]
(d)car t细胞表型的定性评估。显示了来自至少三个独立实验的不同t细胞亚组的平均百分比。psma,前列腺特异性膜抗原;scfv,单链可变片段;efs,延伸因子1alpha短启动子;tn/tscm,幼稚t细胞或记忆t干细胞;tcm,t细胞中央记忆;tem,t细胞效应记忆;teff,t细胞效应器。
[0043]
car t细胞表型的此种表征证实,所选的car设计能够以最少量的终末分化t细胞亚组生成高质量的car t细胞产物。
[0044]
图2.psma-car t细胞的细胞毒性和细胞因子释放。
[0045]
(a)car t细胞对psma阳性c4-2肿瘤细胞的细胞毒性概况。将car t细胞以不同的效应器与靶标(e:t)比率共培养48小时,使用抗原阳性c4-2肿瘤细胞或psma阴性细胞(du145)。
[0046]
(b)car t细胞对psma阴性du 145肿瘤细胞的细胞毒性。将car-t细胞与du145细胞以不同的效应器与靶标(e:t)比率共培养48小时。
[0047]
通过基于xtt elisa的比色测定法测量细胞毒性(%细胞毒性=100-%存活力)。
[0048]
如图2a和图2b中呈现的结果的比较显示,用根据本发明的car转染的t细胞仅对表达psma抗原的细胞系具有细胞毒性活性,因为在图a中,使用表达psma的细胞系(c4-2)。当细胞系不表达psma时,如例如细胞系du145,用car t细胞不能观察到细胞毒性(图2b)。此外,与先前公开的数据(pmid 16204083,pmid:18026115,pmid:19773745,pmid:25358763,pmid:23242161,pmid:4174378,pmid:25279468)相比,本发明的car28和car41细胞显示出优异的细胞毒性:使用1:1的低e:t比率,消除多达90%的psma表达细胞(图2a)。以1:1的低e:t比率观察到的高体外细胞毒性是独特的,到目前为止,尚未在任何可用的psma-car中进行描述,这指示我们基于scfv d7的car t细胞具有优越的性能。
[0049]
(c)定量抗原刺激后促炎细胞因子的释放。用psma阳性c4-2细胞或psma阴性du 145细胞刺激car t细胞48小时。通过基于流式细胞术的细胞因子-珠测定法定量上清液中的细胞因子。***指示统计学显著差异(p<0.001;n=3)。ut;未转导的t细胞,ifn-g;干扰素gamma,gr.a:粒酶a,gr.b;粒酶b。
[0050]
从图2c可以看出,仅当将car t细胞与表达psma的细胞系(细胞系:c4-2)共培养时发生促炎性细胞因子的释放。car28以高于car41细胞的程度被激活。
[0051]
图3.抗原特异性刺激后的car t细胞表型和耗竭概况。
[0052]
(a)car t细胞表型。基于cd62l和cd45ra的表达评估t细胞表型概况之前用psma阳性c4-2肿瘤细胞以1:1效应器与靶标(e:t)比率刺激car t细胞24小时。对于ut对照(未显示)在car-cd3+上,或者对于这两类car t细胞(未显示)在car+cd3+上预门控细胞。
[0053]
(b)car t细胞表型的定量评估。显示了来自三个独立实验的不同t细胞亚组的平均百分比。***(p<0.001)或*(p<0.05)指示统计显著性差异。
[0054]
(c)t细胞耗竭概况。用psma阳性肿瘤细胞(c4-2)刺激car t细胞24小时,然后通过测量cd223(lag-3)和cd279(pd-1)的表达来评估t细胞的耗竭概况。对于ut对照(未显示)在car-cd3+上,或者对于这两类car t细胞(未显示)在car+cd3+上预门控细胞。
[0055]
(d)car t细胞耗竭概况的定量评估。显示了pd-1或lag-3阳性细胞的平均百分比(三个独立的实验)。***指示统计学显著差异(p<0.001)。ns,不显著;ut,未转导的t细胞;tn/tscm,t细胞幼稚或t干细胞记忆;tcm,t细胞中央记忆;tem,t细胞效应记忆;teff,t细胞效应器;lag-3,淋巴细胞激活基因3;pd-1,程序性细胞死亡蛋白1。
[0056]
这组实验解决抗原特异性刺激后t细胞表型以及car41和car28 t细胞的耗竭概况。如在图3a和3b中所见,与car28细胞相反,在抗原特异性刺激后,高百分比的car41细胞保留未分化的t细胞表型。此外,抗原特异性分化后,与car28 t细胞相比,car41细胞对耗竭的敏感性较小(图3c和3d)。
[0057]
图4:前列腺癌靶细胞系c4-2
luc+
的psma表达。
[0058]
对于体内生物发光成像,用编码萤火虫萤光素酶、新霉素抗性基因和绿色荧光蛋白(gfp)的慢病毒载体转导psma阳性的前列腺癌c4-2细胞。转导的细胞系命名为c4-2
luc+
。
[0059]
(a)如通过流式细胞术确定的抗psma mab 3/f11与c4-2
luc+
细胞的结合。
[0060]
(b)如通过流式细胞术确定的在25μg/ml 3/f11的饱和浓度下与c4-2
luc+
和psma阴性du 145细胞的结合。
[0061]
抗psma mab 3/f11,scfv d7的亲本mab的结合验证所有c4-2
luc+
前列腺癌靶细胞表面上的psma表达,而显示du 145前列腺癌对照细胞为psma阴性。
[0062]
图5:如使用活成像(living imaging)系统ivis 200(xenogen vivovision)通过生物发光成像(bli)确定的c4-2
luc+
细胞的生物发光。
[0063]
与萤光素温育后使用体内成像系统ivis 200(xenogen vivovision)测试c4-2
luc+
靶细胞的发光。与未转导的c4-2细胞相比,c4-2
luc+
细胞显示出生物发光,强度(光子/sec/cm2)与细胞数成线性。因此,这些细胞适用于其在小鼠肿瘤异种移植物模型中的用途,通过生物发光成像(bli)检测肿瘤。roi,感兴趣的区域。
[0064]
图6肿瘤内car t细胞疗法
[0065]
(a)肿瘤内car t细胞疗法的示意图。通过皮下注射1.5x106个细胞在动物中建立肿瘤植入。当生长的c4-2
luc+
实体瘤体积达到60-80mm3时,在治疗的第1天以单剂量的5x106个car28(n=5)或car41 t细胞(n=5)肿瘤内注射car t细胞。给对照小鼠注射相同数量的ut细胞(n=7)或保持未治疗(对照,n=6)。通过生物发光成像(bli)显现肿瘤,直到在第22天治疗结束。d,天;i.t.,肿瘤内;s.c.,皮下。
[0066]
(b)肿瘤内注射后car28和car41 t细胞的抗肿瘤活性。在治疗的第1天,给具有皮下生长的c4-2
luc+
实体瘤(60-80mm3体积)的动物肿瘤内注射单剂量的5x106个car28(n=5)或car41 t细胞(n=5)。给对照小鼠注射相同数量的ut细胞(n=7)或保持未治疗(对照,n=6)。通过bli显现肿瘤,直到第22天治疗结束。使用公式vol=d2xd/2从bli图像计算肿瘤体积,以平均值
±
sd表示,其中d为肿瘤的小直径,并且d为肿瘤的大直径。***p<0.001指示统计学显著差异。
[0067]
(c)在治疗结束时(第22天)肿瘤内注射的car28和car41 t细胞的抗肿瘤活性。如上所述,从bli图像计算肿瘤体积。肿瘤体积显示为平均值
±
sd。***p<0.001指示统计学显
著差异。由于对照组的1/6小鼠形成溃疡性肿瘤,因此必须在第15天对此小鼠实施安乐死,并且不将其纳入第22天的统计学分析中。
[0068]
(d)与未治疗的动物(对照)或用ut t细胞治疗的动物相比,在治疗的第1天用单次注射car28或car41 t细胞进行肿瘤内治疗的小鼠的代表性生物发光图像。显示了代表每个治疗组具有大或小肿瘤的动物的两只小鼠。
[0069]
在治疗的第1天,用一次注射5x106个car28 t细胞对携带60-80mm3体积的c4-2
luc+
实体瘤的小鼠进行肿瘤内治疗,导致对肿瘤生长的显著抑制,并且导致5/5完全肿瘤消退(cr),直至第15天(图6b,c和d)。一只动物在第22天显示出肿瘤复发,肿瘤终体积为0.55mm3(图6d,对小鼠拍摄照片)。对于该组的所有小鼠,对第22天计算0.11
±
0.22mm3的平均肿瘤体积(图6b和c)。用car41 t细胞肿瘤内治疗的小鼠显示出不同的响应。注意到1/5cr和1/5部分消退(pr),导致对肿瘤生长的统计学显著抑制,在第22天的平均肿瘤终体积为397.3
±
393.6mm3(图6b和c)。相反,ut组和对照组的所有小鼠均显示出肿瘤进展,导致肿瘤终体积分别为1469.4
±
961.5mm3和1289.1
±
636.8mm3(图6b和c)。
[0070]
图7:在治疗结束时(第22天)肿瘤内治疗的小鼠的体重变化。
[0071]
根据动物保护准则,体重减轻是治疗过程中毒性的重要标志。没有动物显示出超过20%的其初始体重的严重降低。这证明肿瘤内治疗耐受良好,没有明显的脱靶毒性体征。
[0072]
图8:静脉内car t细胞疗法。
[0073]
(a)静脉内car t细胞疗法的示意图。通过皮下注射1.5x106个细胞在动物中建立肿瘤植入。当生长的c4-2
luc+
实体瘤体积达到60-80mm3时,在治疗的第1天,以单剂量的5x10
6 car28(n=5)或car41 t细胞(n=5)静脉内注射car t细胞。给对照小鼠注射相同数量的ut细胞(n=7)或保持未治疗(对照,n=6)。通过bli显现肿瘤直至在第22天治疗结束。d,天;i.v,静脉内;s.c,皮下。
[0074]
(b)静脉内注射后car28和car41 t细胞的抗肿瘤活性。在治疗的第1天,给具有皮下生长的c4-2
luc+
实体瘤(60-80mm3体积)的动物静脉内注射单剂量的5x106个car28(n=5)或car41 t细胞(n=5)。给对照小鼠注射相同数量的未转导的t细胞(ut细胞)(n=5)或保持未治疗(对照,n=6)。通过生物发光成像显现肿瘤,直到第22天治疗结束。使用公式vol=d2xd/2从bli图像计算肿瘤体积,以平均值
±
sd表示,其中d为肿瘤的小直径,并且d为肿瘤的大直径。
[0075]
(c)在治疗结束时(第22天)静脉内注射的car28和car41 t细胞的抗肿瘤活性。如上所述,从bli图像计算肿瘤体积。肿瘤体积显示为平均值
±
sd。
[0076]
(d)静脉内注射单剂量car28或car41 t细胞的小鼠的代表性生物发光图像。显示了代表每个治疗组具有大或小肿瘤的动物的两只小鼠。
[0077]
将car28或car41 t细胞单次静脉内注射到具有60-80mm3体积的实体瘤的小鼠中后,未能测量到肿瘤生长的抑制(图8b和c)。如图8c+d所示,所有治疗组的动物均显示相当的肿瘤生长,导致在第22天肿瘤终体积分别为1469.4
±
961.4mm3(对照),1275.7
±
544.5mm3(ut),1427.3
±
448.5mm3(car28)和1529.3
±
971.0mm3(car41)。
[0078]
图9:组合疗法
[0079]
(a)组合的化学疗法与静脉内car t细胞疗法的示意图。通过皮下注射1.5x106个细胞在动物中建立肿瘤植入。化学疗法在治疗的第1天通过腹腔内注射多西他赛(doc,6mg/
kg bw)开始达3天。在最后一个化学疗法周期后48小时,给小鼠静脉内注射单剂量的5x106个car28(n=3)或car41 t细胞(n=3)。给对照小鼠注射单独的多西他赛(n=3)或保持未治疗(对照,n=4)。通过bli显现肿瘤,直到在第17天治疗结束。d,天;i.v,静脉内注射;i.p,腹膜内;s.c,皮下。
[0080]
(b)与多西他赛化学疗法组合的car28和car41 t细胞的抗肿瘤活性。在治疗的第1、2和3天,对具有皮下生长的c4-2
luc+
肿瘤(200
–
250mm3)的小鼠腹腔内注射多西他赛(doc;6mg/kg bw)。48小时后,再对小鼠静脉内注射单剂量的5x106个car28(n=3)或car41 t细胞(n=3)。通过bli监测肿瘤的生长。与未治疗的对照(n=4)相比,doc治疗可显著抑制肿瘤生长,其通过添加car28或car41 t细胞得到增强。对照组的4/4小鼠在治疗期间显示出生长中的肿瘤,而在doc组中,2/3小鼠显示出pr。在doc+car28组中,1/3的动物显示pr。在doc+car41组中,2/3小鼠显示cr,并且1/3小鼠显示pr。doc+car28和doc+car41组之间的统计学显著差异证明了该治疗方案中car41 t细胞的优异效果。*p<0.05指示统计学显著差异。
[0081]
(c)在治疗的第17天,与多西他赛化学疗法组合的car28和car41 t细胞的抗肿瘤活性。在第17天治疗结束时,测定对照组小鼠的平均肿瘤体积为1817.5
±
165.5mm3。doc组和doc+car28组分别表现出338.3
±
355.6mm3和559.9
±
317.2mm3的显著更低的体积。doc+car41组的动物仅具有50.2
±
71.1mm3的平均肿瘤体积。*p<0.05指示统计学显著差异。
[0082]
(d)用doc预治疗并静脉内注射单剂量car28或car41 t细胞的小鼠的代表性生物发光图像。显示了代表每个治疗组具有大或小肿瘤的动物的两只小鼠。
[0083]
总之,用多西他赛化学疗法对肿瘤进行的预治疗导致静脉内应用的car41 t细胞的抗肿瘤活性。
[0084]
图10:序列
[0085]
图10显示了源自小鼠的抗原结合构建体d7的氨基酸序列(“框1”)。此外,提供了编码核酸序列及其互补链。cdr h1-h3和cdr l1-l3核酸和氨基酸序列用灰色箭头标记。
[0086]
d7的scfv片段的氨基酸序列(seq id no:1)对于本发明而言是必不可少的,因为它是抗原结合片段的人源化的起始序列。人源化序列与seq id no:1具有至少80%的同源性。在更优选的实施方案中,序列与seq id no:1具有至少90%,且更优选至少95%的同源性,并且甚至更优选与seq id no:1具有至少98%的同源性。应当指出,图10中所示的cdr区在很高水平上是保守的,这意味着通过kabat方法确定的cdr区具有不超过三个,优选地不超过一个,并且优选地没有氨基酸交换。
[0087]
本申请公开的序列概括如下:
[0088]
[0089][0090]
图11:人源化序列中的潜在突变
[0091]
在图11中显示了cdr。通常,cdr-h1,cdr-h3和cdr-l3没有任何修改。但是,可以替换cdr-l2中的一个氨基酸,替换cdr-l1中的多达两个氨基酸,以及cdr-h3中的多达六个氨基酸。可以替换的氨基酸称为x1-x9,其含义如图11的图例所示。
[0092]
图12:构建体car28的序列
[0093]
图12a-12d显示了称为car-cd28的构建体的序列。
[0094]
图13:构建体car41的序列
[0095]
在图13a-13d中显示了构建体car-4-1bb的序列。
[0096]
应当注意,在图10-13中,序列信息与序列的相关部分具有的功能的信息一起提供。因此,应当理解,本领域技术人员从所提供的序列中以最佳方式获得信息。序列10-13公
开了本发明的优选实施方案。
[0097]
图14:源自d7的人源化scfv序列
[0098]
(a)显示了d7的重链的鼠序列及其五个人源化的变体,其中根据kabat确定以及也根据imgt,显示了cdr-h1,cdr-h2和cdr-h3的位置。
[0099]
(b)显示了具有cdr-l1,cdr-l2和cdr-l3的d7轻链的鼠序列及其五个人源化变体。应当理解,重链和轻链可以彼此组合。例如,可以将变体1的重链序列(人d7-vh1)与变体5的轻链(人d7-vl5)组合。
[0100]
图15:根据本发明的car构建体与其中的psma抗原结合片段已经在现有技术中描述的构建体的比较
[0101]
(a)表达第二代抗-psma car的自我失活逆转录病毒载体的示意图。car表达由efs启动子驱动。抗psma car含有与fc igg1衍生的铰链区,跨膜(tm)域,cd28衍生的共刺激域和源自cd3ζ链的胞内信号传导域融合的单链可变片段(scfv)3d8或j591(均在现有技术中进行了描述)或d7(源自针对psma的3f/11鼠单克隆抗体)。铰链区提供了scfv和tm域之间的物理间隔区,以实现最佳靶标识别。cd28共刺激域含有氨基酸交换,其在抑制性调节性t细胞(treg)存在的情况下阻止lck结合并增强抗肿瘤活性。所有产生的逆转录病毒构建体具有相同的支架,但仅在scfv片段上不同,从而允许car构建体在活性和细胞毒性方面进行并排比较。
[0102]
(b)用编码三种不同psma-car的逆转录病毒颗粒转导jurkat细胞中的car表达。病毒转导后,将jurkat细胞扩充16天,然后收获细胞,并且用抗人igg抗体(car)染色以评估转导效率和car表达水平。
[0103]
(c)抗原特异性激活概况,用psma阳性,pd-l1阴性的c4-2肿瘤细胞(psma+/pdl1-)或psma阳性,pd-l1阳性lncap肿瘤细胞(psma+/pdl1+)以1:1的效应器与靶标比率刺激表达car的jurkat细胞24小时。作为阴性对照,将car细胞与psma阴性du145肿瘤细胞(psma-)共培养。刺激24小时后,收获细胞,并通过评估激活标志物cd69阳性细胞的百分比来评估激活概况。
[0104]
将基于d7的car t jurkat细胞与基于j591和基于3d8的car t jurkat细胞并排比较抗原特异性激活概况。如图c所示,基于d7的car和基于j591的car能够在抗原特异性致敏后介导jurkat细胞的大规模激活,如通过在约-70%的细胞中激活标志物cd69的上调所测量的。激活不受抑制性配体pd-l1的存在的影响。另一方面,基于3d8的car t细胞仅被弱激活(多至20%的cd69阳性细胞)。
[0105]
(d)在原代t细胞中的car表面表达。在逆转录病毒转导之前,用抗cd2/cd3/cd28抗体将t细胞激活2-3天。扩充6-9天后,收获细胞并用抗人igg抗体(car)和cd3染色,以评估转导效率和car表达水平。
[0106]
(e-g)通过用抗cd3/cd28/cd2抗体激活3天后转导原代t细胞来生成car t细胞的细胞毒性概况。扩充8-9天后,通过以不同的效应器与靶标(e:t)比率,c4-2(psma+/pdl1-)肿瘤细胞(e),lncap(psma+/pdl1+)肿瘤细胞(f)或du145(psma-)肿瘤细胞(g),共培养car t细胞48小时来检查细胞毒性概况。通过基于xtt elisa的比色测定法测量细胞毒性(%细胞毒性=100%-%存活力)。*(p<0.05),**(p<0.01)或***(p<0.001)指示统计学显著差异。ut,未转导的细胞;psma,前列腺特异性膜抗原;pd-l1,程序性细胞死亡配体1。
[0107]
将基于d7的car t细胞与基于j591的car t细胞并排比较。与基于j591的car t细胞相比,基于d7的car t细胞在两个肿瘤细胞靶标(e,f)上均显示出优异的细胞毒性概况,如通过这两个肿瘤细胞系都可以以较低的效应器与靶标比率消除的实情证明的。由于较弱的激活概况(c)和t细胞中有效表达的缺乏(d),在比较中不进一步包括基于3d8的car t细胞。总之,基于d7的car t细胞的psma抗原特异性激活和细胞毒性概况两者均优于基于现有技术的car的car t细胞。
[0108]
(h,i)使用的前列腺癌细胞系的表征。在c4-2,lncap和du145前列腺细胞系上评估psma靶抗原表达(h)和pd-l1(cd274)表达(i)的程度。对于流式细胞仪分析,将细胞用3/f11抗体(抗psma)或抗cd274抗体染色。ut,未转导的t细胞;pd-l1,程序性细胞死亡配体1;psma,前列腺特异性膜抗原。
[0109]
如图h所示,c4-2和lncap肿瘤细胞均表达psma抗原,而du145为psma抗原阴性。图i显示lncap和du145细胞的大部分表达pd-l1,而c4-2细胞对pd-l1呈阴性。
[0110]
图中显示的实验结果可以如下解释:
[0111]
通常,car由含有识别抗原的scfv的胞外域,铰链区,跨膜区和一个或多个激活t细胞的胞内信号传导域(包括cd3ζ链)组成。在第二代或第三代car中,包括通常源自cd28,4-1bb,ox40,cd27和/或icos的共刺激域(图1a)。为了治疗前列腺癌,许多尝试已经利用靶向psma表位的car,并且这些中的一些策略已经进入临床试验(例如nct01140373,nct01929239)。然而,这些car的效力在体外和体内似乎都相当低。特别地,以基于抗psma scfv 3d8或j591(称为pzi)的第一代car为基础的早期研究显示,所产生的car t细胞的低效力,如通过必须使用高达100:1的高效应器与靶标(e:t)比率在体外消除肿瘤细胞的需要指示。当使用基于d2b或j591衍生的scfv的第二代或第三代car时,体外效力得到改善。但是,在异种移植肿瘤小鼠模型中,这些car t细胞的效力仍然较低,如通过尽管应用很高的car t细胞剂量,有时高达20x106个car t细胞,或多次输注,但是这些靶向psma的car t细胞仅能抑制肿瘤的生长,但不能在体内消除肿瘤的实情(pmid 16204083,pmid:18026115,pmid:19773745,pmid:25358763,pmid:23242161,pmid:4174378,pmid:25279468)指示。鉴于与相当低效的psma-car t细胞的这些混合结果,旨在基于scfv d7生成并验证新型、更有效的靶向psma的car。设计了两种不同的第二代psma-car,它们带有cd28(car28)或4-1bb(car41)衍生的共刺激域(图1)。结果证明这些新开发的基于d7的psma靶向性car t细胞在体外和体内均具有高的有效性。制备的car t细胞产品包含高百分比的未分化t细胞,例如幼稚t细胞、t干细胞记忆和中央记忆t细胞表型(图1d)。这些基于d7的car t细胞在体外以低的效应器与靶标比率完全消除psma阳性肿瘤细胞,并在特异性抗原刺激后释放预期的细胞因子(图2a-c)。
[0112]
与car28相比,在抗原特异性刺激后,car41 t细胞维持更为幼稚的表型和更少的耗竭概况(图3)。
[0113]
重要的是,在肿瘤内应用后,基于d7的car t细胞消除小鼠肿瘤模型中的psma阳性实体瘤(图6)。可以看出,对照(未治疗)和具有未转导(ut)t细胞的对照形成快速生长的肿瘤。然而,本发明的两个实施方案(car28 t细胞和car41 t细胞)明显地阻断肿瘤内单剂量施用后的肿瘤生长并导致pr或cr。这证明了该构思在体内起作用。
[0114]
本干预的car t细胞的静脉内单剂量注射没有引起对psma阳性实体瘤的生长抑制
(图8)。但是,在用多西他赛的化学疗法后,单剂量静脉内注射的car41 t细胞导致大肿瘤(200-250mm3)的完全消退(图9)。
[0115]
将基于d7的car t细胞与基于j591和基于3d8的car t细胞进行了并排比较(图15)。在用编码基于d7,j591和3d8的car的表达载体转导的jurkat细胞中比较了抗原特异性激活概况。尽管基于d7的car和基于j591的car能够在抗原特异性致敏后介导jurkat细胞的大规模激活(图15c),但带有3d8-car的细胞仅被弱激活。此外,在原代t细胞转导后,与基于j591的car t细胞相比,基于d7的car t细胞在两种psma肿瘤细胞系上显示出优异的细胞毒性概况(图15e,15f),如以较低的效应器与靶标比率消除这两种细胞系的实情证明。
[0116]
总之,已证明基于d7的抗psma car t细胞具有出乎意料的特性,所述特性优于先前发布的靶向psma的car t细胞,特别是考虑到其优异的体外细胞毒性和高体内抗肿瘤活性。因此,它们是开发用于治疗晚期前列腺癌的新型免疫疗法的有前途的工具。
[0117]
因此,本发明涉及用于t细胞的嵌合抗原受体,其包含特异性结合psma抗原的抗原结合片段。抗原结合片段优选包含用合适的接头连接的v
h
和v
l
片段。此外,嵌合抗原受体优选包含间隔元件,跨膜片段和cdr3ζ胞质域。此外,嵌合抗原受体优选包含来自cd28(图12)和/或4-1bb(图13)胞质域的片段。
[0118]
本发明的嵌合抗原受体优选含有至少三个选自cdr-h1,cdr-h2,cdr-h3,cdr-l1,cdr-l2和cdr-l3的cdr,如图10所示。cdr由氨基酸序列和相关编码核酸序列上方的灰色箭头显示。在一个优选的实施方案中,嵌合抗原受体包含分别如图10和11所示的cdr的至少三个(cdr-h1,cdr-h3,cdr-l3),优选四个(cdr-h1,cdr-h3,cdr-l2,cdr-l3),并且更优选为至少五个(cdr-h1,cdr-h3,cdr-l1,cdr-l2,cdr-l3)。
[0119]
嵌合抗原受体优选以人源化形式存在。此类人源化形式可通过将至少三个,优选四个,更优选五个或六个cdr插入与如图11所示的鼠支架具有高度同源性的合适的人抗原结合支架中来获得。
[0120]
通常,在合适的载体的帮助下将编码嵌合抗原受体的遗传信息引入靶免疫细胞(例如t细胞)中。此类载体可以优选是慢病毒载体或逆转录病毒载体或转座子或质粒。或者,如本文所述,可以在设计者核酸酶技术例如crispr/cas技术或talen技术的帮助下以靶向方式将遗传信息引入t细胞的基因组中。
[0121]
在另一方面,本发明涉及提供包含如本文所述的嵌合抗原受体的t细胞的体外方法。第一步,优选通过白细胞单采术方法从供体分离t细胞。这导致t细胞的大量富集。然后,分别通过用合适的载体转染或通过含有嵌合抗原受体的遗传信息的病毒载体转导来对t细胞进行遗传修饰。然后可以分离并扩增此类经遗传修饰的t细胞,从而可以将不含期望遗传信息的那些t细胞与经修饰的t细胞分离或至少减少。然后可以将转染的t细胞应用于待治疗的患者。
[0122]
实施例1
[0123]
car编码逆转录病毒颗粒的制备
[0124]
于37℃在具有5%co2的潮湿培养箱中将hek293t细胞在补充有10%胎牛血清(biochrom,berlin,germany),青霉素(100u/ml),链霉素(100mg/l)和10mm hepes(sigma-aldrich)的dmem(gibco,invitrogen,karlsruhe,germany)中培养。转染前一天,将细胞以5x106个细胞/皿的细胞密度接种到10cm皿中。24小时后,使用聚乙烯亚胺(pei:0.1mg pei/
ml,polysicence inc.,usa)转染细胞。每10cm皿使用3μg编码vsv-g包膜的质粒,6μg gag/pol编码质粒和10μg编码car28或car41的载体质粒。转染后48和72小时,收集含有病毒载体的上清液,并使用超速离心(wx ultra系列;thermo scientific:25,000rpm达2小时,于4℃)浓缩。将浓缩的载体悬浮于100μl的冷pbs中,并保持在-80℃直至使用。通过转导jurkat t细胞系,然后用抗人igg染色转导的细胞来确定car阳性细胞,从而确定载体制剂的生物效价。
[0125]
实施例2
[0126]
靶向psma的car t细胞的生成
[0127]
从外周血单个核细胞(pbmc)产生car t细胞。使用相分离法(ficoll,sigma-aldrich)根据制造商的建议分离pbmc,然后在液氮中冷冻直至使用。为了产生car t细胞,将pbmc解冻,并在rpmi完全培养基[补充有10%胎牛血清(biochrom,berlin,germany),青霉素(100u/ml),链霉素(100mg/l)和10mm hepes缓冲液(sigma-aldrich)的rpmi 1640培养基(gibco,invitrogen,karlsruhe,germany)]中恢复24小时。然后,使用抗cd2/cd3/cd28抗体(immunocult,stemcell technologies)激活pbmc,并用补充有100u/ml il-2、25u/ml il-7和50u/ml il-15(均来自miltenyi biotech)的rpmi完全培养基培养2至3天,之后用编码car28或car41的gamma-逆转录病毒构建体以范围为50-300的moi转导。将转导的细胞在用聚d-溶素(pdl,sigma-aldrich)包被的孔中培养,该孔含有补充有5μg/ml的鱼精蛋白硫酸盐(sigma-aldrich)和1000u/ml的il-2,25u/ml的il-7和50u/ml的il-15的rpmi完全培养基。一天后,更换培养基,并在补充有100u/ml il-2、25u/ml il-7和50u/ml il-15的rpmi完全培养基中将细胞进一步扩充8-9天,然后在液氮中冷冻直至进一步使用。
[0128]
实施例3
[0129]
car t细胞的质量评估
[0130]
为了通过流式细胞术(facs canto ii或accuri,bd biosciences)监测car和tcr的表达,分别用抗人igg-pe(southern biotech)和cd3-apc(miltenyi biotec)对细胞进行染色。如图1b所示,car28或car41均达到高达50%的转导效率。为了进行质量评估,收获car t细胞并在扩充期结束时用抗人cd62l-bv421(bd biosciences),抗人cd45ra-fitc(biolegend),抗人cd3-apc/h7(bd biosciences)和抗人igg染色-pe(car)(southern biotec)染色(图1c,d)。基于cd62l和cd45ra的表达确定t细胞表型。对于未转导的(ut)t细胞在cd3+/car-上或者对于这两类car t细胞在cd3+/car+上预门控细胞(图1c,d)。转导和扩充方案均未诱导t细胞分化,因为存在高百分比的具有幼稚t细胞(tn),t干细胞记忆(tscm)或中央记忆t细胞(tcm)表型的未分化细胞与低分数的终末分化的效应t细胞(teff)的组合(图1c,d),指示该方案允许生成高质量的car t细胞产物。
[0131]
实施例4
[0132]
制备的car t细胞的体外细胞毒性
[0133]
如先前所述,通过使用xtt测定法评估细胞存活力来确定制备的car t细胞的细胞毒性潜力。将car t细胞与psma阳性c4-2肿瘤细胞或抗原阴性肿瘤对照细胞(du145)在96孔板中以不同的效应器与靶标比率在最终体积200μl/孔的没有任何细胞因子的rpmi完全培养基中共培养48小时。为了确定细胞存活力作为代谢活性的功能,取出100μl/孔的培养基,并用100μl/孔的xtt溶液(sigma-aldrich)代替,并在37℃温育细胞。使用elisa读数器
rpe(becton-dickinson,mountain view,ca)在冰上温育40分钟。然后重复洗涤细胞,并悬浮于200μl含有1μg/ml碘化丙啶,3%fcs和0.1%nan3的pbs中。使用带有cellquest pro软件的facscalibur流式细胞仪(bd biosciences,heidelberg,germany)进行染色细胞的分析。
[0145]
如图4a所示,mab 3/f11以0.96μg/ml(约6.3nm)的结合常数(kd)与c4-2
luc+
细胞结合,该结合常数定义为半最大饱和浓度。这证明了在此细胞系上psma的高表达。在饱和浓度下,c4-2
luc+
细胞显示为psma阳性,而du 145前列腺癌对照细胞显示为psma阴性(图4b)。
[0146]
实施例8
[0147]
c4-2
luc+
细胞的生物发光
[0148]
借助于体内成像系统ivis 200(xenogen vivovision)测试c4-2
luc+
细胞的生物发光。为此,将不同数量的细胞接种到96孔板(nunc delta surface,thermo fisher scientific,roskilde,denmark)中,并与40μl萤光素/孔(biosynth ag,staad,switzerland)作为底物一起温育。使用软件活成像(living image)3.0在10-30分钟内(caliper lifesciences)分析发光。
[0149]
生物发光成像(bli)显示,c4-2
luc+
细胞的数量与生物发光之间存在线性关系(量化为感兴趣区域(roi)。用未转导的c4-2细胞不能检测到发光信号,并且背景染色以最大2.362x106个光子/sec/cm2的roi确定(图5)。
[0150]
实施例9
[0151]
通过肿瘤内注射car t细胞治疗c4-2
luc+
肿瘤异种移植物
[0152]
向5-6周龄的雄性scid cb17/lcr-prkdc scid/crl小鼠(20-25g,janvier labs,st berthevin cedex,france)注射与50%matrigel(collaborative biomedical products,chicago,il)混合的pbs溶液中的1.5x106个c4-2
luc+
个细胞,s.c.注射到右体侧中。通过bli监测肿瘤的生长。为此,将200μl的萤光素(biosynth ag,staad,switzerland)i.p.注射到动物中,并且注射后使用活成像系统ivis 200(xenogen vivovision)在麻醉下进行bli达10-30分钟。使用软件活成像3.0和公式体积=d2xd/2从bli照片计算肿瘤体积,其中d是肿瘤的大直径,并且d是肿瘤的小直径。当肿瘤达到约60-80mm3的体积时,分别向小鼠肿瘤内注射仅一剂5x106个car28(n=5)或car41 t细胞(n=5)(治疗第1天)(图6a)。作为对照,给小鼠注射未转导的t细胞(ut,n=7)或保持未治疗(对照,n=6)。在治疗的第1、3、8、15和22天通过bli监测肿瘤的生长。对照组(ut和未治疗)显示快速的肿瘤生长。用car41 t细胞达到减少的肿瘤生长。在用car28 t细胞治疗的小鼠中发现了明显的肿瘤消退(图6b,c和d)。直到第22天治疗结束,对照组的所有动物均显示出生长的肿瘤。根据动物保护准则,未治疗组的一只小鼠不得不在第15天因溃疡性肿瘤而被处死。
[0153]
在第22天,分别达到平均肿瘤终体积1469.4
±
961.5mm3(未治疗的对照)和1289.1
±
636.8mm3(ut)。相反,用car28 t细胞治疗的5/5小鼠从第8天起显示完全的肿瘤消退(cr),并且4只小鼠保持无肿瘤,直到实验结束。在剩下的动物中,在第22天检测到小肿瘤,并通过组织学检查证实为0.55mm3的体积。对于用car28治疗的所有小鼠,在第22天计算平均肿瘤体积0.11
±
0.22mm3,与对照组相比具有统计学显著性(*p<0.05)(图6b和6d)。肿瘤内注射car41 t细胞的小鼠对治疗显示不同响应。治疗的五只动物之一显示cr(图6b和6d)。另一只动物显示pr,而其他动物直到治疗的第22天都具有肿瘤进展。总之,确定该组的平均
肿瘤体积为397.7
±
393.6mm3(图6c)。
[0154]
实施例10
[0155]
不良副作用
[0156]
根据动物保护准则,观察到小鼠在治疗期间明显的毒性体征(体重减轻和食欲不振,毛皮变化,发烧,紧张,漠然,攻击,呼吸系统病症,瘫痪,死亡),并且终止标准定义如下:大量减少食物摄入,初始体重降低>20%,痉挛,瘫痪,异常呼吸病症,漠然,攻击性作为严重疼痛的体征,肿瘤直径>20mm,溃疡性肿瘤。除未治疗的对照组的一只小鼠因溃疡性肿瘤不得不在第15天处死外,没有小鼠显示明显的毒性体征,并且也没有动物达到一种或多种测定标准。图7表明,没有动物显示>20%的其初始体重的严重降低。总之,所有动物都已经完全耐受用抗psma car t细胞的肿瘤内治疗,没有明显的非特异性毒性体征。
[0157]
实施例11
[0158]
通过静脉内注射car t细胞治疗c4-2
luc+
肿瘤异种移植物
[0159]
将具有c4-2
luc+
肿瘤异种移植物的小鼠分别静脉内注射100μl pbs中的仅一剂5x106个car28(n=5),car41(n=5)或ut t细胞(n=5)(治疗第1天)(图8a)。对照小鼠(n=6)保持未治疗。如上所述监测肿瘤的生长。与对照组(肿瘤终体积:1469.4
±
961.5mm3)和ut组(肿瘤终体积:1275.7
±
544.5mm3)相比,注射car28 t细胞的小鼠在第22天治疗结束时显示相似的肿瘤生长,平均肿瘤体积为1427.3
±
448.5mm3(图8b和8c)。对于用car41 t细胞治疗的动物,确定1529.3
±
971.0mm3的平均肿瘤体积(图8c)。静脉内用car41或car28治疗的动物中的肿瘤生长不受控制(图8b,c和d)。
[0160]
总之,肿瘤内应用psma car t细胞(car28和car41两者)在体内完全消除肿瘤(图6)。相反,静脉内注射car t细胞不导致任何抗肿瘤活性(图8),这表明肿瘤内注射允许car t细胞绕过tme,而静脉内注射后car t细胞可能受到tme的抑制。
[0161]
实施例12
[0162]
通过化学疗法和静脉内注射car t细胞的组合治疗c4-2
luc+
肿瘤异种移植物
[0163]
对具有大c4-2
luc+
肿瘤异种移植物(200-250mm3)的小鼠腹膜内注射3个每日周期的多西他赛(doc,在治疗的第1、2和3天时6mg/kg bw)(图9a)。在最后一个周期后48小时,分别给小鼠静脉内注射仅一剂5x106个car28(n=3)car41 t细胞(n=3)或pbs(n=3)(图9a)。对照动物(n=4)保持完全未治疗。如上所述,通过bli监测肿瘤的生长。与对照组相比,注射单独的doc和doc+car28的小鼠显示对肿瘤生长的抑制(图9b)。与对照组(1817.5
±
165.5mm3)相比,在第17天达到仅用doc治疗的小鼠的肿瘤终体积338.2
±
355.6mm3和用doc+car28治疗的小鼠的肿瘤终体积599.9
±
317.2mm3(图9c)。相反,用doc+car41组合疗法治疗的2/3动物显示cr,并且1/3小鼠显示pr(图9b)。测定50.2
±
71.0mm3的平均肿瘤体积(图9c)。
[0164]
这些结果指示,d7-car t细胞在单剂量施用car t细胞组合化学治疗剂如多西他赛后有效消除大肿瘤(图9d)。
[0165]
实施例13
[0166]
鼠序列的人源化
[0167]
为了避免以后潜在的不利影响,例如在人类患者中产生的针对抗原结合片段的鼠部分的抗体,将源自构建体d7的抗原结合片段人源化。
[0168]
在使序列人源化的过程中,将源自构建体d7的抗psma结合序列与人干细胞序列进
行了比较。选择具有很高同源性的序列,并且已经确定了cdr结合序列所在的区域。为了确定cdr序列,已知几种方法。最优选的方法是根据kabat的方法和imgt方法。imgt(international immunogene tics数据库)是专门研究免疫球蛋白分子的高质量综合信息系统。由于根据kabat和imgt的方法提供略有不同的结果,因此图14a和b中显示了不同变体的序列(人d7vh1-5和人d7 vl1-5)。显示根据kabat和imgt的相关cdr区。
[0169]
重要的是要注意,即使通过使用两种不同的方法,根据这两种确定方法,具有氨基酸序列ardgnfpyyamds(seq id no:11)的cdrh3的序列和具有序列sqsthvpt(seq id no:12)的cdrl3的序列也是相同的。因此,假定那两个cdr,即cdrh3和cdrl3对于构建体的适当功能是重要的。如图14a和b所示的人源化序列对应于seq id no:13至23。
[0170]
实施例14
[0171]
已经将根据本发明的优选构建体的生物活性与其中psma结合片段源自抗体j591的构建体进行了比较。此外,将其与其中抗原结合构建体源自另一种抗体(3d8)的相似构建体进行比较。
[0172]
在图15a-15i中描述实验和从中获得的结果。
[0173]
图15a是除psma结合片段以外相同的不同抗psma-car28构建体的示意图。在构建体car28(d7)中,使用根据本发明的片段。在car28(j591)中,psma片段源自抗体j591,而在car28(3d8)中,psma片段源自psma结合抗体(3d8)。
[0174]
将构建体引入jurkat t细胞系中,并测量构建体的表达。在图15b中,对照称为ut(未转导)。图15b显示除称为ut(未转导)的对照外,在流式细胞仪中测量的所有car构建体均在jurkat t细胞系中正确表达。
[0175]
在图15c中,通过监测抗原刺激后激活标志物的表达来测量转导的jurkat细胞的抗原特异性激活概况。与基于d7和j591的car相反,基于3d8的car在扩充成psma表达性肿瘤细胞后仅介导转导的jurkat细胞的弱激活。
[0176]
图15d显示了在原代t细胞中的car表达。可以看出,与car28(j591)(30%)相比,根据本发明的构建体car28(d7)被更好地表达(42%)。其中psma结合片段源自3d8的构建体的表达非常低(4%)。在原代t细胞中未以充分程度表达的构建体不适合用于以后的治疗用途。
[0177]
图15e-15g中显示了car28 t细胞对指示细胞即c4-2(psma+/pdl1-);lncap(psma+/pdl1+)和du145(psma-/pdl1+)的细胞毒性概况。为了评估car t细胞的细胞毒性概况,已经确定了不同比率的效应器(car28 t细胞):靶细胞(不同的肿瘤细胞,即c4-2,lncap和du145)。从图15e和图15f可以看出,根据本发明的构建体优于具有源自构建体j591的抗原结合片段的构建体。在较低的效应器:靶标比率下,根据本发明的构建体总是优于对比构建体。效应器靶标比是临床使用的非常重要的方面,因为在现实生活中无法提供很高的效应器:靶标比率。效应器:靶标比率越低,构建体越好。
[0178]
图15h和图15i显示了前列腺癌细胞系lncap,c4-2和du145上的psma和pd-l-1表达。
[0179]
实施例15
[0180]
在另一个实验中,可以显示化学疗法与car疗法的组合优于单独的化学疗法。
[0181]
在图16a和图16b中显示了实验。图16a是用多西他赛(doc)加car t细胞对携带皮
下c4-2
luc
异种移植物的小鼠的组合疗法的示意图。在第1天和第2天给具有150-200mm3肿瘤的小鼠注射2剂6mg/kg bw doc,接着在第8天i.v.注射car28或car41 t细胞。通过触诊和bli监测肿瘤生长直至第22天。
[0182]
在图16b中显示实验结果:car28和car41 t细胞与doc组合在携带皮下c4-2
luc
异种移植物的小鼠中的体内抗肿瘤活性。在治疗的第15、17和22天确定了doc+car28治疗的动物与未治疗的对照之间的肿瘤体积的统计学显著差异。在治疗的第17和22天确定了用doc+car28治疗的动物和仅用doc治疗的小鼠之间的肿瘤体积的统计学显著差异。此外,在第22天,用doc+car41治疗的动物的肿瘤体积显著小于对照组。这证明组合治疗优于单独的化学疗法。未配对t检验,*p<0.05。
[0183]
从实验中可以清楚地看出,根据本发明的car28和car 41构建体与化学治疗剂一起具有协同作用,其中该作用已经用多西他赛例举。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1