生产吡唑化合物的方法与流程
本发明涉及通过新型中间体生产取代的吡唑基化合物的方法和使得能够通过商业可行的合成路线合成最终产物的用于制备这类中间体的独特方法。本发明还涉及生产取代的吡唑基甲状腺样化合物的新型方法,和固体形式的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸、其药物组合物及其制备方法。
背景技术:
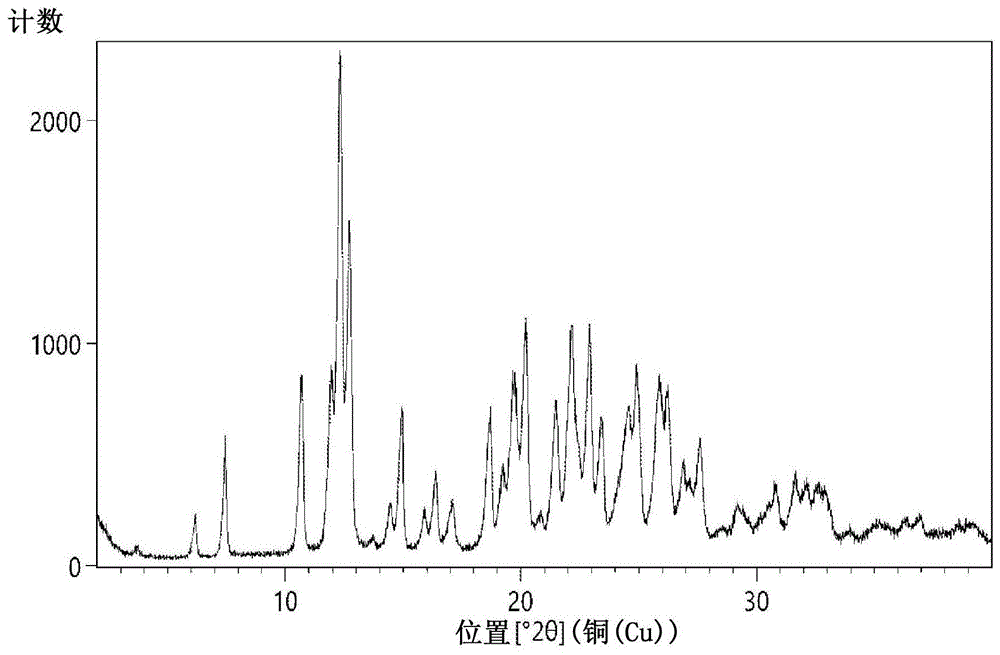
:在美国专利no.8,143,424和8,378,118中公开了取代的吡唑基甲状腺样化合物。这些专利中所述的合成路线包括用碘甲烷保护中间体的羟基,随后进行后续合成转化。最终,用三溴化硼使甲基保护基脱保护以获得最终产物。在性质上,碘甲烷和三溴化硼是高危险性的。因此,在本领域中需要提供避免使用这些试剂以及还适合于商品化生产的生产取代的吡唑基甲状腺样化合物的替代方法。此外,上述美国专利中描述的用于生产中间体的合成方法包括危险性试剂,如锌-汞和氯化铝的使用。所述方法还包括在高温下的反应、高真空蒸馏和用于纯化的柱色谱法,由于其消耗更多的溶剂、时间和能量,这使得药物产品合成在商业规模上是不可行的。因此,本发明人已开发了新型合成路线,其避免了以上缺点并且通过商业上可行的合成路线生产最终产物。本文还描述了固体形式的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。与其作为药物的效力相关的固体的性质可以依赖于固体的形式。例如,在药物物质中,固体形式的变化可以导致性质差异,如溶解速率、口腔吸收、生物利用度、毒理学结果以及临床效力。本文描述的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的固体形式是3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式、盐、溶剂化物和水合物。技术实现要素:本发明涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或c1-20烷基三环(三环烷基,alkyltricyclic);n是1至5;所述方法包括:用适合的保护基保护化学式(ii)的化合物以获得化学式(iii)的化合物,其中,r1如上所述;其中r1为如上所定义的并且pg1是羟基保护基,但条件是pg1不是甲基;用适合的还原剂还原化学式(iii)的化合物以获得化学式(iv)的化合物,其中以上定义了r1和pg1;将化学式(iv)的化合物与适合的羟基活化剂反应以获得化学式(v)的化合物,其中r1和pg1为如上所定义的,并且x是离去基团;将化学式(v)的化合物转化为化学式(vi)的化合物,其中r1、r2、r3和pg1为如上所定义的;使化学式(vi)的化合物与化学式(vii)或(viib)的化合物反应以获得化学式(viii)的化合物,其中r4和n为如上所定义的,并且x是离去基团;其中r1、r2、r3、r4、n和pg1为如上所定义的;并且使化学式(viii)的化合物脱保护以获得化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;x是cl、br或i;并且pg1是苄基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或c1-20烷基三环;n是1至5;所述方法包括将化学式(v)的化合物转化为化学式(via)的化合物,其中r1为如上所定义的并且pg1是羟基保护基,但条件是pg1不是甲基并且x是离去基团;其中r1、r2、r3和pg1为如上所定义的;使化学式(via)的化合物与肼反应以获得化学式(vi)的化合物,其中r1、r2、r3和pg1为如上所定义的;并且将化学式(vi)的化合物转化为化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;x是cl;并且pg1是苄基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或c1-20烷基三环;n是1至5;所述方法包括用适合的保护基保护化学式(ix)的化合物以获得化学式(x)的化合物,其中r1为如上所定义的;其中r1为如上所定义的并且pg1是羟基保护基,但条件是pg1不是甲基;将化学式(x)的化合物转化为化学式(v)的化合物,其中r1和pg1为如上所定义的,并且x是离去基团;并且将化学式(v)的化合物转化为化学式(i)的取代的吡唑化合物。[或]用适合的保护基保护化学式(ii)的化合物以获得化学式(iii)的化合物,其中r1如上所描述的;其中r1为如上所定义的并且pg1是羟基保护基,但条件是pg1不是甲基;使化学式(iii)的化合物与还原剂反应以获得化学式(iv)的化合物,其中r1和pg1是以上所定义的;使化学式(iv)的化合物与适合的羟基活化剂反应以获得化学式(v)的化合物,其中r1和pg1为如上所定义的,并且x是离去基团;并且将化学式(v)的化合物转化为化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;x是cl;并且pg1是苄基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或c1-20烷基三环;n是1至5;所述方法包括当r4为c1-10饱和或不饱和的烷基、c1-10取代的或未取代的烷基芳基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或者c1-20烷基三环时,水解化学式(viiia)的化合物以获得化学式(xi)的化合物;其中r选自h或pg1;其中r、r1、r2、r3、n为如上所定义的并且pg1是羟基保护基,但条件是pg1不是甲基;其中r、r1、r2、r3、n和pg1为如上所定义的并且r5为h;并且任选地使化学式(xi)的化合物脱保护以获得化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;x是cl;并且pg1是苄基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或c1-20烷基三环;n是1至5;所述方法包括将化学式(iii)的化合物转化为化学式(xii)的化合物,其中r1为如上所定义的并且pg1是羟基保护基,但条件是pg1不是甲基;其中r1、r2、r3和pg1为如上所定义的;还原化学式(xii)的化合物以获得化学式(via)的化合物,其中r1、r2、r3和pg1为如上所定义的;使化学式(via)的化合物与肼反应以获得化学式(vi)的化合物,其中r1、r2、r3和pg1为如上所定义的;并且将化学式(vi)的化合物转化为化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;并且pg1是苄基。本发明还涉及用于产生化学式(ix)的化合物的方法,其中r1选自h、c1-6烷基、c1-6烷氧基和卤素;所述方法包括用适合的保护基保护化学式(xiii)的化合物以获得化学式(xiv)的化合物,其中r1为如上所定义的;其中r1为如上定义的并且pg2是选自甲苯磺酰基或苯甲酰基的羟基保护基;使化学式(xiv)的化合物与2,2-二甲基-1,3-二噁烷-4,6-二酮反应以获得化学式(xv)的化合物,其中r1和pg2为如上所定义的;使化学式(xv)的化合物环化以获得化学式(xvi)的化合物,其中r1和pg2为如上所定义的;使化学式(xvi)的化合物脱保护以获得化学式(xvii)的化合物,其中r1为如上所定义的;并且还原化学式(xvii)的化合物以获得化学式(ix)的化合物。在优选的实施方式中,取代基r1是甲基。本发明还涉及用于产生化学式(ix)的化合物的方法,其中r1选自h、c1-6烷基、c1-6烷氧基和卤素;所述方法包括用适合的保护基保护化学式(xiii)的化合物以获得化学式(xiv)的化合物,其中r1为如上所定义的;其中r1为如上定义的并且pg2是选自甲苯磺酰基或苯甲酰基的羟基保护基;使化学式(xiv)的化合物与2,2-二甲基-1,3-二噁烷-4,6-二酮反应以获得化学式(xv)的化合物,其中r1和pg2为如上所定义的;使化学式(xv)的化合物环化以获得化学式(xvi)的化合物,其中r1为如上所定义的;还原化学式(xvi)的化合物以获得化学式(xvia)的化合物,其中r1和pg2为如上所定义的;并且使化学式(xvia)的化合物脱保护以获得化学式(ix)的化合物,在优选的实施方式中,取代基r1是甲基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基、环亚酰胺或c1-20烷基三环;n是1至5;所述方法包括使化学式(vi)的化合物与化学式(viia)或(viic)的化合物反应以获得化学式(xviii)的化合物,其中r1、r2、r3和pg1为如上所定义的;其中r6选自单羧酸基或二羧酸基、酰基氯基、酯基、腈基、醛基、醇基、酰胺基、烷基、烯基、炔基、卤代烷基或酮基;x是离去基团;并且n为如上所定义的;其中r1、r2、r3、r6、n和pg1为如上所定义的;任选地将化学式(xviii)的化合物转化为化学式(viii)的化合物,其中r1、r2、r3、r4、n和pg1为如上所定义的;并且将化学式(viii)的化合物脱保护以获得化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;r6是酯;n为2;并且pg1是苄基。本发明还涉及新型中间体化合物,其在化学式(i)的取代的吡唑化合物的制备中是有用的。本发明的新型中间体化合物包括化学式(iii)、(v)、(vi)、(via)、(viii)和(xii)所表示的化合物。其中r1、r2、r3、r4、x、n和pg1为如上所定义的。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;并且pg1是苄基。本发明涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的新方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基或c1-20烷基三环;n是1至5;所述方法包括使化学式(xix)的化合物与化学式(xx)的化合物在存在缩合剂的情况下缩合以获得化学式(viiia)的化合物,其中r2、r3、r4和n为如上所定义的;其中r是h或pg1;其中pg1是羟基保护基,并且r1为如上所定义的;其中r、r1、r2、r3、r4和n为如上所定义的;并且任选地使化学式(viiia)的化合物脱保护和/或水解以获得化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;并且pg1是甲基或苄基。在一个实施方式中,通过所述方法产生化学式(xix)的化合物,其中r2、r3、r4和n为如上所定义的;所述方法包括使化学式(xxi)的化合物与化学式(vii)的化合物缩合以获得化学式(xix)的化合物,其中r2、r3、r4和n为如上所定义的;其中r4和n为如上所定义的,并且x是离去基团。在优选的实施方式中,取代基r2和r3是甲基;r4是h或c1-6烷基;n为2;并且x是cl或br。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的新方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基或c1-20烷基三环;n是1至5;所述方法包括使化学式(xx)的化合物与化学式(xxii)的化合物在存在缩合剂的情况下缩合以获得化学式(vib)的化合物,其中r是h或pg1,其中pg1是羟基保护基,并且r1为如上所定义的;其中r2和r3为如上所定义的;其中r、r1、r2和r3为如上所定义的;使化学式(vib)的化合物与肼环化以获得化学式(vic)的化合物,其中r、r1、r2和r3为如上所定义的;并且将化学式(vic)的化合物转化为化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;并且pg1是甲基或苄基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的新方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基或c1-20烷基三环;n是1至5;所述方法包括使化学式(xx)的化合物与化学式(xxiii)的化合物在存在缩合剂的情况下缩合以获得化学式(xxiv)的化合物,其中r是h或pg1;其中pg1是羟基保护基,并且r1为如上所定义的;其中r1和r3为如上所定义的,并且r5是h或pg3;其中pg3是氨基保护基;其中r、r1、r2、r3和r5为如上所定义的;任选地将化学式(xxiv)的化合物脱保护以获得化学式(vic)的化合物,其中r、r1、r2和r3为如上所定义的;并且将化学式(vic)的化合物转化为化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;并且pg1是甲基或苄基。本发明还涉及生产化学式(i)的取代的吡唑化合物或其药学上可接受的盐、溶剂化物、共晶体和水合物的新方法:其中:r1、r2和r3各自独立的选自h、c1-6烷基、c1-6烷氧基或卤素;r4选自h、c1-10饱和或不饱和的烷基、c1-10取代的或未取代的芳基烷基、c3-10环烷基、芳基、杂芳基、c1-10烷基甲硅烷基、c1-10烷基甲硅烷基醚、取代的或未取代的卤代烷基或c1-20烷基三环;n是1至5;所述方法包括使化学式(xxv)的化合物与化学式(xx)的化合物在存在缩合剂的情况下缩合以获得化学式(xviiia)的化合物,其中r2、r3和n为如上所定义的,并且r6选自单羧酸基或二羧酸基、酰基氯基、酯基、腈基、醛基、醇基、酰胺基、烷基、烯基、炔基、卤代烷基或酮基;其中r是h或pg1,其中pg1是羟基保护基,并且r1为如上所定义的;其中r、r1、r2、r3、r4、r6和n为如上所定义的;任选地将化学式(xviiia)的化合物转化为化学式(viiia)的化合物,其中r、r1、r2、r3、r4和n为如上所定义的;并且任选地使化学式(viiia)的化合物脱保护和/或水解以获得化学式(i)的取代的吡唑化合物。在优选的实施方式中,取代基r1、r2和r3是甲基;r4是h或c1-6烷基;n为2;并且pg1是甲基或苄基。在一个实施方式中,通过所述方法产生化学式(xxv)的化合物,其中r2、r3、r6和n为如上所定义的;所述方法包括使化学式(xxi)的化合物与化学式(viia)的化合物缩合以获得化学式(xxv)的化合物,其中r2和r3为如上所定义的;其中r6和n为如上所定义的,并且x是离去基团;在优选的实施方式中,取代基r2和r3是甲基;r6是单羧酸或酯;n为2;并且x是cl或br。本发明还涉及制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的方法。所述方法包括用酸处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的酯以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。本发明还提供了包含治疗有效量的化学式(i)的取代的吡唑化合物和一种或多种药物可用的赋形剂和/或载体的药物组合物。本发明还提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的多晶型形式。在一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1。在一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1显示出具有在约6.1±0.2、7.4±0.2、10.7±0.2、11.9±0.2、12.3±0.2、12.7±0.2、14.9±0.2、18.7±0.2、20.2±0.2、22.1±0.2和22.9±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1的特征在于与图1所提供的xrpd图基本类似的x射线粉末衍射(xrpd)谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1的特征在于与图2中的dsc热谱图基本类似的差示扫描量热法(dsc)热谱图(温谱图,thermogram)。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1的特征在于与图3中的tga曲线基本类似的热重分析(tga)曲线。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1的方法。所述方法包括:a)用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯,和b)在约10℃至约50℃用酸使步骤a)的反应混合物酸化以获得结晶形式-t15-1。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1的方法。所述方法包括:a)在存在碱水溶液的情况下,用适合的羟基脱保护剂处理3-(4-{[7-(pg1-氧)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸或其酯,其中pg1是羟基保护基,和b)在约10℃至约50℃用酸使步骤a)的反应混合物酸化以获得结晶形式-t15-1。在另一个实施方式中,本发明提供了包含治疗有效量的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1和药物可用的赋形剂和/或载体的药物组合物。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2。在一个实施方式中,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2,其显示出具有在约7.4±0.2、11.9±0.2、12.7±0.2和14.9±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2的特征还在于具有在约12.2±0.2、18.7±0.2、19.7±0.2、22.1±0.2、24.8±0.2和25.8±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2的特征在于与图4所提供的x射线粉末衍射谱图基本类似的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2的特征在于与图5中的dsc热谱图基本类似的差示扫描量热法(dsc)热谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2的特征在于与图6中的tga曲线基本类似的热重分析(tga)曲线。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2的方法。所述方法包括:a)用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯,b)在10℃以下使步骤a)的反应混合物酸化,c)分离所述固体,和d)使所述固体在真空下干燥以提供结晶形式-t15-2。在另一个实施方式中,本发明提供了包含治疗有效量的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2和药物可用的赋形剂和/或载体的药物组合物。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3。在一个实施方式中,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3,其显示出具有在约6.2±0.2、10.7±0.2、16.5±0.2和21.6±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的特征还在于具有在约12.4±0.2、20.3±0.2、22.5±0.2和23.0±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的特征在于与图7中所提供的x射线粉末衍射谱图基本类似的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的特征在于与图8中的dsc热谱图基本类似的差示扫描量热法(dsc)热谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的特征在于与图9中的tga曲线基本类似的热重分析(tga)曲线。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的方法。所述方法包括:在有机溶剂或有机溶剂和水的混合物中用碱处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯,然后在10℃以上用酸酸化以提供结晶形式-t15-3。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的方法。所述方法包括在10℃以上,在有机溶剂或有机溶剂和水的混合物中用适合的羟基脱保护剂处理3-(4-{[7-(pg1-氧)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸,其中pg1是羟基保护基;以获得结晶形式-t15-3。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3的方法。所述方法包括将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸与有机溶剂结合,和任选地添加抗-溶剂以提供3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3。在另一个实施方式中,本发明提供了包含治疗有效量的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3和药物可用的赋形剂和/或载体的药物组合物。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4。在一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4是水合物。在另一个实施方式中,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4,其显示出具有在约8.1±0.2、11.9±0.2、12.6±0.2和19.6±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4的特征在于与图10所提供的x射线粉末衍射谱图基本类似的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4的特征在于与图11中的dsc热谱图基本类似的差示扫描量热法(dsc)热谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4的特征在于与图12中的tga曲线基本类似的热重分析(tga)曲线。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4的方法。所述方法包括:a)用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯,b)在10℃以下用酸使步骤a)的反应混合物酸化,c)分离所述固体,和d)空气干燥所述固体以提供结晶形式-t15-4。在另一个实施方式中,本发明提供了包含治疗有效量的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4和药物可用的赋形剂和/或载体的药物组合物。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的二甲基亚砜(dmso)溶剂化物。在一个实施方式中,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物,其显示出具有在约13.2±0.2、14.9±0.2、16.7±0.2、18.5±0.2和22.4±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物的特征在于与图13所提供的x射线粉末衍射谱图基本类似的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物的特征还在于与图14中的dsc热谱图基本类似的差示扫描量热法(dsc)热谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物的特征还在于与图15中的tga曲线基本类似的热重分析(tga)曲线。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物的方法。所述方法包括用dmso处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸以提供3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物。在另一个实施方式中,本发明提供了包含治疗有效量的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物和药物可用的赋形剂和/或载体的药物组合物。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐。在一个实施方式中,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐,其显示出具有在约13.9±0.2、14.4±0.2、19.5±0.2、21.7±0.2和22.5±0.2以2-θ度所表示的特征峰的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐的特征在于与图16所提供的x射线粉末衍射谱图基本类似的x射线粉末衍射谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐的特征还在于与图17中的dsc热谱图基本类似的差示扫描量热法(dsc)热谱图。在另一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐的特征还在于与图18中的tga曲线基本类似的热重分析(tga)曲线。在另一个实施方式中,本发明提供了制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的二钠盐的方法。所述方法包括在有机溶剂、水或它们的混合物中用钠离子源处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸以提供3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐。在另一个实施方式中,本发明提供了包含治疗有效量的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐和药物可用的赋形剂和/或载体的药物组合物。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的无定形形式。在另一个方面,本发明提供了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的无定形固体分散体。在另一个方面,本发明提供了治疗受试者中的疾病的方法,其包括以下步骤:施用治疗有效量的药物组合物,其包含3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1或形式-t15-2或形式-t15-3或形式-t15-4或dmso溶剂化物或者二钠盐或者无定形形式和药物可用的赋形剂和/或载体。附图说明在所附方面具体描述了本发明的新颖特征。通过参考描述其中使用了本发明的原理的说明性实施方式的以下详细说明和附图,将获得对本发明的特征和优势的更好理解,其中:图1示出了结晶形式-t15-1的x射线粉末衍射(xrpd)图。图2示出了结晶形式-t15-1的差示扫描量热法(dsc)热谱图。图3示出了结晶形式-t15-1的热重分析(tga)曲线。图4示出了结晶形式-t15-2的xrpd图。图5示出了结晶形式-t15-2的dsc热谱图。图6示出了结晶形式-t15-2的tga曲线。图7示出了结晶形式-t15-3的xrpd图。图8示出了结晶形式-t15-3的dsc热谱图。图9示出了结晶形式-t15-3的tga曲线。图10示出了结晶形式-t15-4的xrpd图。图11示出了结晶形式-t15-4的dsc热谱图。图12示出了结晶形式-t15-4的tga曲线。图13示出了dmso溶剂化物的xrpd图。图14示出了dmso溶剂化物的dsc热谱图。图15示出了dmso溶剂化物的tga曲线。图16示出了二钠盐的xrpd图。图17示出了二钠盐的dsc热谱图。图18示出了二钠盐的tga曲线。图19示出了无定形形式的xrpd图。具体实施方式尽管本文已显示和描述了本发明的所选实施方式,但是对于本领域技术人员显而易见的是仅通过举例说明来提供这些实施方式。在不背离本发明的情况下,本领域技术人员现将能够想到多种变化、改变和替换。应理解本文描述的本发明的实施方式的多种替代可以用于实践本发明。除非另外受限于特殊情况,否则如在整个说明书中所使用的,以下定义适用于术语。除非上下文另外明确规定,否则如本文所使用的,单数形成的“一”、“一个”和“该”包括多个对象。如本文所使用的术语“约”是指指示值的数值的±10%。如本文所使用的,术语“适合的保护基”或“羟基保护基”是指暂时封闭化合物中羟基反应性位点的部分。通常,这样做是为了在多官能性化合物中的另一个反应性位点选择性进行化学反应或稳定羟基。优选地,羟基保护基是通过化学反应选择性可除去的。“适合的保护基”或“羟基保护基”包括形成酯、醚或甲硅烷基-保护基的基团。例如,所形成的酯是乙酰基(ac)、苯甲酰基(bz)或者新戊酰基(piv)。例如,所形成的醚保护基是甲基、苄基(bn)、甲氧基乙氧基甲醚(mem)、三苯甲基(tr)、对甲氧苯甲基(pmb)、二甲氧基三苯甲基(dmt)、甲氧基甲醚(mom)、四氢吡喃基(thp)或者乙氧基乙醚(ee)。例如,所形成的甲硅烷基保护基是三甲基甲硅烷基(tms)、三乙基甲硅烷基(tes)、叔丁基二甲基甲硅烷基(tbdms或tbs)、三-异丙基甲硅烷基氧基甲基(tom)或者三异丙基甲硅烷基(tips醚)。所形成的其它保护基是对溴苯甲酰基、对硝基苯甲酰基、三异丙基甲硅烷基(tips)、苄氧甲基(bom)、p-甲氧基-苄氧甲基(pmbm)、[(3,4-二甲基苄基)氧]-甲基(dmbm)、甲硫基甲基(mtm)、2-(三甲基-甲硅烷基)-乙氧基甲基(sem)。术语“溶剂”包括单一溶剂或溶剂混合物,如水、酯、醇、卤代烃、酮、醚、极性非质子溶剂、烃熔剂或它们的混合物。酯的实例包括乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸叔丁酯和乙酸正丁酯。醇的实例包括具有1至6个碳原子的伯、仲和叔醇。醇的实例包括甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧基乙醇。卤代烃的实例包括二氯甲烷、氯仿和1,2-二氯乙烷。酮的实例包括丙酮、甲基异丁基酮和甲基乙基酮。醚的实例包括二乙醚、2-甲基四氢呋喃、环戊基甲醚和四氢呋喃。极性非质子溶剂的实例包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮。烃熔剂的实例包括c6至c7取代的或未取代的非环或环脂肪烃、己烷、庚烷、环己烷和芳烃,如甲苯、二甲苯、苯。根据本发明的“离去基团”包括(但不限于)卤代基团,具体地氟代、氯代、溴代或碘代,和取代的或未取代的烷基磺酰氧基、取代的或未取代的芳基磺酰氧基、取代的或未取代的芳基烷基磺酰氧基和取代的或未取代的卤代烷基磺酰氧基。根据本发明的术语“还原剂”是指导致有机分子还原反应的试剂。适合的还原剂的实例包括氢化物还原剂、硼烷和金属催化剂。优选的氢化物还原剂是硼氢化钠、三乙酰氧基氢硼化钠、硼氢化锂、二异丁基氢化铝、氢化铝锂、三乙基硅烷等;优选的硼烷包括处于回流四氢呋喃中的乙硼烷和硼烷等。金属催化剂是作为元素或处于盐的形式并且纯的或处于惰性载体上的铜、钯、铂、镍、锌、铁、铑、钌。适合的催化剂包括雷尼-镍、碳负载钯(palladiumoncarbon)、碳负载铂(platinumoncarbon)等。优选的金属催化剂选自雷尼-镍、碳负载钯、碳负载铂、碳负载钌(rutheniumoncarbon)、碳负载铑(rhodiumoncarbon)和二氧化铂,具体地选自雷尼-镍、碳负载钯、碳负载铂和二氧化铂。根据本发明的术语“酸催化剂”可以是路易斯酸或质子酸。例如,路易斯酸是氯化锌、碘化锌、氯化铝、溴化铝和三氟化硼醚化物。质子酸(例如)包括无机酸,如盐酸、氢溴酸、硝酸、硫酸和磷酸、羧酸,如乙酸和三氟乙酸、磺酸,如甲磺酸、苯磺酸、1-萘磺酸、1-丁烷磺酸、乙烷磺酸、4-乙基苯磺酸、1-己烷磺酸、1,5-萘磺酸、1-辛烷磺酸、樟脑磺酸、三氟甲烷-磺酸和p-甲苯-磺酸。另外,术语“酸催化剂”包括阳离子交换树脂,其还可以被称为树脂-基酸催化剂。根据本发明的术语“羟基脱保护试剂”是指能够切割羟基保护基以形成游离羟基的试剂。这些羟基脱保护剂的实例包括(但不限于)三溴化硼、四-正-丁基氟化铵、三(二甲基氨)二氟三甲基硅酸锍、溴化氢、氟化氢、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、三氟乙酸、对甲苯磺酸吡啶盐、p-甲苯磺酸、甲酸、高碘酸、碳负载钯和碱。根据本发明的术语“羟基活化剂”是指提高羟基反应性,借此使羟基成为更好的离去基团的试剂。这些羟基活化剂的实例包括p-甲苯磺酰氯、4-硝基苯磺酰氯、亚硫酰氯、草酰氯、三氟甲磺酸酐、甲磺酰氯、甲磺酸酐、三苯基膦、酰基氯、4-二甲基氨吡啶、三烷基膦、三芳基膦、三杂芳基-膦、三甲基膦、三丁基膦、三(o-甲苯基)膦、三(m-甲苯基)膦、三(p-甲苯基)膦、三(2-吡啶基)-膦、三(3-吡啶基)-膦或三(4-吡啶基)膦。根据本发明的术语“酸”包括无机酸或有机酸。无机酸包括盐酸、氢溴酸、硫酸、硝酸和磷酸等。有机酸包括甲酸、乙酸、丙酸、丁酸、戊酸、己酸、草酸、乳酸、苹果酸、柠檬酸、碳酸、琥珀酸、苯甲酸和p-甲苯磺酸等。根据本发明的术语“碱”包括碱金属碳酸盐,如碳酸钠、碳酸钾、碳酸锂等;碱金属酸式碳酸盐,如碳酸氢钠、碳酸氢钾等;碱金属氢氧化物,如氢氧化钠、氢氧化钾、氢氧化锂等;碱金属醇化物,如甲醇钠、乙醇钠、甲醇钾、乙醇钾、叔丁醇钠、叔丁醇钾、叔丁醇锂等;碱金属氢化物,如氢化钠、氢化钾、氢化锂等;碱金属酰胺,如氨基钠、氨基钾、氨基锂等;和有机碱,如二甲胺、二乙胺、二异丙胺、二异丙基乙胺、二异丁胺、三乙胺、吡啶、4-二甲基氨吡啶、n-甲基吗啉、2,6-二甲基吡啶、二异丙基氨基锂;有机硅碱,如六甲基二硅氮烷锂、六甲基二硅氮烷钠、六甲基二硅氮烷钾或它们的混合物。如本文所使用的术语“氨基保护剂”是指暂时阻断化合物中的胺反应性位点的部分。通常,这样做是为了在多官能性化合物中的另一个反应性位点选择性进行化学反应或稳定氨基。优选地,氨基保护基是通过化学反应选择性可除去的。“氨基保护基”包括9-芴基甲基氨基甲酸酯、甲酰基、乙酰基、三氟乙酰基、苄氧羰基、苯甲酰基、苄基、苄氧羰基、m-硝基苯氧基羰基、o-硝基苄氧羰基、3,5-二甲氧基苄氧基-羰基、p-硝基苄氧羰基、p-甲氧苯甲基、p-甲氧基苄基羰基、3,4-二甲基苄基、叔丁氧羰基、三甲基甲硅烷基、甲苯磺酰基、2-三甲基甲硅烷基乙烷-磺酰基、二苯甲基、三氯乙基氯甲酸酯、三苯甲基和取代的三苯甲基、烯丙氧基羰基、硝基藜芦基氧基羰基和烯丙氧基羰基。如本文所使用的术语“氨基脱保护剂”是指将选择性除去氨基保护基的试剂或试剂组合。在特定反应中使用的脱保护剂的选择将取决于其上发生反应的化合物的保护基及其它特征。脱保护剂的实例包括(但不限于)试剂,如盐酸、氢溴酸、硫酸、甲酸、三氟乙酸、碳负载钯或它们的混合物。术语“结合”包括添加、溶解、制浆、搅拌或它们的组合。术语“治疗有效量”是指足以实现预期应用的本文描述的化合物的量,所述预期应用包括(但不限于)疾病治疗,如以下所定义的。基于预期应用或要治疗的受试者和疾病状况,例如,受试者的体重和年龄、疾病状况的严重程度、施用方式等(这些是本领域的技术人员可以容易地确定的),治疗有效量可以是不同的。该术语还适用于将在靶细胞中引起特定反应的剂量,例如,血小板粘附和/或细胞迁移减少。基于所选的具体化合物、要遵循的剂量施用方案、是否与其它化合物组合施用、施用时机、所施用的组织和携带它的物理递送系统,具体的剂量将是不同的。根据本发明的术语“相转移催化剂”包括季铵盐、鏻盐(phosphoniumsalt)、冠醚、聚乙二醇和它们的组合。季铵盐包括(例如)四丁基卤化铵、四丁基硫酸氢铵、四丁基硫氰酸铵、四丁基四氟硼酸铵、苄基三丁基卤化铵、十六烷基三甲基卤化铵、十六烷基三甲基硫酸氢铵、甲基三乙基卤化铵、甲基三(十八基)卤化铵、四乙基卤化物铵、六氟磷酸四乙基铵、四乙基四氟硼酸铵、四己基硫酸氢铵、四甲基卤化铵、四辛基卤化铵及它们的混合物。鏻盐包括(例如)三丁基十六基磷鎓和四丁基磷鎓卤化物及它们的混合物。冠醚包括(例如)12-冠-4、1-氮杂-15-冠-5、18-冠-6、二苯并-18-冠醚-6、二环己并-18-冠-6、二环己并-24-冠-8、三[2-(2-甲氧乙氧基)乙基]胺、4,7,13,6,21,24-六氧杂-1,10-二氮杂双环[8.8.8]二十六烷及它们的混合物。聚乙二醇包括(例如)聚乙二醇200、400、600、1000及它们的混合物。如本文所使用的,术语“治疗效果”是指如上所述的治疗益处和/或预防益处。预防益处包括疾病或病况出现的延迟或消除、疾病或病况症状发病的延迟或消除、疾病或病况发展的减缓、停止或逆转或它们的任意组合。根据本发明的术语“药物可用的赋形剂”或“药物可用的载体”表示(但不限于)处于适合的剂量形式的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的组合物所需的任何一种或多种非活性成分。具体地,所述赋形剂包括(但不限于)稀释剂、载体、填充剂、膨胀剂、粘结剂、崩解剂、聚合物、润滑剂、助流剂、表面活性剂、稳定剂、吸收加速剂、调味剂、防腐剂、抗氧化剂、缓冲剂、释放修饰材料、涂层材料和制药工业中常用的任何其它赋形剂。术语“受试者”是指动物,如哺乳动物,例如人。本文描述的方法可以用于人治疗剂和兽医应用两者。在一些实施方式中,所述患者是哺乳动物,并且在一些实施方式中,所述患者是人。实验仪器和条件使用panalyticalx射线衍射仪,使用cukα-辐射进行x射线粉末衍射(xrpd)。标准测量条件是:管能:45kv/40ma;步长:0.008°(20);脉冲时间:10±2sec;扫描范围:2°-40°(20);发散狭缝:自动;样品旋转;使用x-celerator检测器;y轴显示强度值并且x轴显示2-θ值。使用mettlertoledo差示扫描量热仪2,使用密封的铝坩埚并且以10℃min-1的加热速率在25℃至300℃的范围进行差示扫描量热法(dsc)。使用mettlertoledo-tg1,使用氧化铝坩埚,在氮气氛下并且以10℃min-1的加热范围,在25℃至300℃的范围内进行热解重量分析(tga)。使用brukertensor27仪,在4000cm-1至400cm-1的范围内,以2cm-1的分辨率使用20次扫描进行傅里叶变换红外光谱学(ftir)。在本发明的一个具体方面,在存在碱和活化剂的情况下,在溶剂中用适合的保护剂保护化学式(ii)的化合物的羟基以获得化学式(iii)的化合物。优选地,所述适合的保护基是苄基。优选地,在存在碱,如氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的情况下进行羟基保护。更优选地,所述碱是碳酸钾。优选地,所述活化剂是(例如)碘化钾。优选地,在存在极性非质子溶剂,如n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮的情况下进行羟基保护。更优选地,所述极性非质子溶剂是n,n-二甲基甲酰胺。可以通过如现有技术,如(例如)us8,378,118中已知的方法,从化学式(ix)的化合物制备化学式(ii)的化合物。然后,在溶剂中在存在催化剂的情况下用还原剂处理化学式(iii)的羟基保护的化合物以提供化学式(iv)的化合物,通过在存在溶剂的情况下,使化学式(iv)的化合物与羟基活化剂反应将其进一步转化为化学式(v)的化合物。优选地在存在还原剂,如硼氢化钠、三乙酰氧基氢硼化钠、硼氢化锂、二异丁基氢化铝、氢化铝锂的情况下进行还原。更优选地,所述还原剂是硼氢化钠。优选地,在存在催化剂,如,例如,乙酸的情况下进行还原。优选地,在存在卤化溶剂,如,例如,二氯甲烷的情况下进行还原。优选地,在存在羟基活化剂,如p-甲苯磺酰氯、4-硝基苯磺酰氯、亚硫酰氯、草酰氯、三氟甲磺酸酐、甲磺酰氯、甲磺酸酐或酰基氯的情况下,进行化学式(iv)的化合物向化学式(v)的化合物的转化。更优选地,所述羟基活化剂是亚硫酰氯。优选地,在存在溶剂,如,例如,二氯甲烷的情况下进行羟基激活反应。然后,在存在碱和活化剂的情况下,在溶剂中使化学式(v)的化合物与乙酰丙酮反应。在存在酸催化剂的情况下,在溶剂中用肼进一步处理所得产物(即,化学式(via)的化合物)以获得化学式(vi)的化合物。优选地,所述碱选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾。更优选地,所述碱是碳酸钾。优选地,所述活化剂是(例如)碘化钠或碘化钾。优选地,在存在溶剂,如,例如,丙酮和催化的量的n,n-二甲基甲酰胺的情况下进行化学式(v)的化合物向化学式(via)的化合物的转化。所述酸催化剂优选地选自乙酸或三氟乙酸。更优选地,所述酸催化剂是乙酸。优选地,在存在选自甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行化学式(via)的化合物与肼的反应。优选地,所述溶剂是异丙醇。然后,在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱的情况下,在溶剂中使化学式(vi)的化合物与化学式(vii)的化合物缩合以获得化学式(viii)的化合物。更优选地,所述碱是碳酸铯。优选地,在存在极性非质子溶剂,如(例如)n,n-二甲基甲酰胺的情况下进行缩合反应。可以在存在铜催化剂,例如,氯化铜(ii)或硫酸铜(ii)的情况下进行化学式(vi)的化合物与化学式(viib)的化合物的反应。在溶剂中,用羟基脱保护剂进行化学式(viii)的化合物的羟基脱保护以获得化学式(i)的取代的吡唑化合物。优选地,所述羟基脱保护剂是(例如)碳负载钯。优选地,在存在氢气压力的情况下进行脱保护反应。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。优选地,在存在选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行羟基脱保护。更优选地,所述溶剂是甲醇或乙酸乙酯。根据本发明的另一个方面,在存在碱和活化剂的情况下,在溶剂中用适合的保护基保护化学式(ix)的化合物的羟基以获得化学式(x)的化合物。优选地,所述适合的保护基是苄基。优选地,在存在碱,如氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的情况下进行羟基保护。更优选地,所述碱是碳酸钾。优选地,所述活化剂是(例如)碘化钠或碘化钾。优选地,在存在极性非质子溶剂,如n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮的情况下进行羟基保护。更优选地,所述极性非质子溶剂是n,n-二甲基甲酰胺。然后,在存在盐酸和酸催化剂的情况下,在溶剂中用甲醛或多聚甲醛处理化学式(x)的保护的化合物以获得化学式(v)的化合物。优选地,在存在选自氯化锌、碘化锌、氯化铝、溴化铝、硫酸、磷酸、乙酸或三氟乙酸的酸催化剂的情况下进行反应。更优选地,所述酸催化剂是氯化锌、乙酸或它们的混合物。优选地,在存在选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯和乙酸正丁酯的溶剂的情况下进行化学式(x)的化合物与甲醛或多聚甲醛的反应。更优选地,所述溶剂是乙酸乙酯。通过如上描述的方法进行化学式(v)的化合物向化学式(i)的取代的吡唑化合物的转化。在本发明的另一个方面,在存在碱的情况下通过酯水解进行化学式(viiia)的化合物的水解以提供化学式(xi)的化合物。优选地,通过在溶剂中使化学式(viii)的化合物与选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱反应进行酯水解以获得化学式(xi)的化合物。更优选地,所述碱是氢氧化钠。优选地,在存在溶剂,如水、四氢呋喃或它们的混合物的情况下进行反应。在溶剂中,用羟基脱保护剂进行化学式(xi)的化合物的脱保护以获得化学式(i)的取代的吡唑化合物。优选地,所述羟基脱保护剂是(例如)碳负载钯。优选地,在存在氢气压力的情况下进行脱保护反应。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。优选地,在存在选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行羟基脱保护。更优选地,所述溶剂是甲醇或乙酸乙酯。在本发明的另一个方面,在存在催化剂的情况下,在溶剂中,化学式(iii)的化合物与乙酰丙酮反应以获得化学式(xii)的化合物。所述催化剂优选地选自哌啶、乙酸或它们的混合物。优选地,所述溶剂是甲苯。优选地,在约30℃至约所述反应混合物的回流温度的温度下进行化学式(iii)的化合物与乙酰丙酮的反应。在存在催化剂的情况下用适合的还原剂进一步还原化学式(xii)的化合物以提供化学式(via)的化合物。所述还原剂和催化剂分别优选地选自锌和乙酸。优选地,在约0℃至50℃的温度下进行还原剂对化学式(xii)的化合物的还原。在存在酸催化剂的情况下,在溶剂中用肼进一步处理化学式(via)的化合物以获得化学式(vi)的化合物。所述酸催化剂优选地选自乙酸或三氟乙酸。更优选地,所述酸催化剂是乙酸。优选地,在溶剂中在约20℃至约90℃进行化学式(via)的化合物与肼的环化。更优选地,所述反应在约60℃至约85℃进行。优选地,在存在选自甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行化学式(via)的化合物与肼的反应。更优选地,所述溶剂是异丙醇。在本发明的另一个方面,通过在存在碱和活化剂的情况下,在溶剂中用适合的保护基保护化学式(xiii)的化合物来制备化学式(ix)的化合物以获得化学式(xiv)的化合物。优选地,所述适合的保护基是甲苯磺酰基。优选地,在存在碱,如氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的情况下进行羟基保护。更优选地,所述碱是碳酸钾。优选地,所述活化剂是(例如)碘化钠或碘化钾。优选地,在存在极性非质子溶剂,如n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮的情况下进行羟基保护。更优选地,所述极性非质子溶剂是n,n-二甲基甲酰胺。可以通过现有技术中已知的方法,如(例如)通过如us8,486,996中所述的方法或者通过如本文描述的方法从2-甲酚制备化学式(xiii)的化合物。在存在碱和催化剂的情况下,在溶剂中进行化学式(xiv)的化合物与2,2-二甲基-1,3-二噁烷-4,6-二酮(米氏酸(meldrum’sacid))的反应以获得化学式(xv)的化合物。优选地,在存在选自吡啶、三甲胺、三乙胺、二乙胺、二异丙基乙胺、咪唑、三乙醇胺、吗啉或n-甲基吗啉的碱的情况下进行反应。更优选地,所述碱是三乙胺。优选地,在存在催化剂,如,例如,甲酸的情况下进行反应。此外,优选地,在存在极性非质子溶剂,其选自n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮的情况下进行反应。更优选地,所述极性非质子溶剂是n,n-二甲基甲酰胺。通过在溶剂中使化学式(xv)的化合物与卤化剂,然后与路易斯酸反应来进行化学式(xv)的化合物的环化以获得化学式(xvi)的化合物。所述卤化剂优选地为(例如)亚硫酰氯。所述路易斯酸优选地选自氯化锌、碘化锌、氯化铝、溴化铝和三氟化硼醚化物。更优选地,所述路易斯酸是氯化铝。所述溶剂可以优选地选自甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇、2-乙氧乙醇、二氯甲烷、氯仿、1,2-二氯乙烷或它们的混合物。更优选地,所述溶剂是二氯甲烷、甲醇或它们的混合物。在存在碱的情况下,在溶剂中,进行化学式(xvi)的化合物的脱保护以获得化学式(xvii)的化合物。优选地,在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠或碳酸氢钾的碱的情况下进行脱保护反应。更优选地,所述碱是氢氧化钠。优选地,在存在选自水、甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇、2-乙氧乙醇或它们的混合物的溶剂的情况下进行脱保护反应。更优选地,所述溶剂是水、甲醇或它们的混合物。在存在选自硼氢化钠、三乙酰氧基氢硼化钠、硼氢化锂、二异丁基氢化铝、氢化铝锂、三乙基硅烷和碳负载钯的还原剂的情况下进行化学式(xvii)的化合物的还原。更优选地,所述还原剂是碳负载钯。优选地,在存在氢气压力的情况下进行还原。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。优选地,在存在催化剂,如,例如,乙酸的情况下进行还原。优选地,在存在醇溶剂,如(例如)甲醇、乙醇和异丙醇的情况下进行还原。更优选地,所述溶剂是甲醇。在存在选自硼氢化钠、三乙酰氧基氢硼化钠、硼氢化锂、二异丁基氢化铝、氢化铝锂、三乙基硅烷和碳负载钯的还原剂的情况下进行化学式(xvi)的化合物的还原。更优选地,所述还原剂是三乙基硅烷。优选地,在存在催化剂,如(例如)氯化铝或三氟化硼二乙醚的情况下进行还原。优选地,在存在醇溶剂,如(例如)甲醇、乙醇和异丙醇的情况下进行还原。更优选地,所述溶剂是甲醇。在存在碱的情况下,在溶剂中,进行化学式(xvia)的化合物的脱保护以获得化学式(ix)的化合物。优选地,在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠或碳酸氢钾的碱的情况下进行脱保护反应。更优选地,所述碱是氢氧化钠。优选地,在存在选自水、甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇、2-乙氧乙醇或它们的混合物的溶剂的情况下进行脱保护反应。更优选地,所述溶剂是水、甲醇或它们的混合物。在本发明的另一个方面,在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱的情况下,在溶剂中进行化学式(vi)的化合物与化学式(viia)的化合物的反应以获得化学式(xviii)的化合物。优选地,在存在极性非质子溶剂,如(例如)n,n-二甲基甲酰胺的情况下进行反应。任选地在存在选自氢醌、铁催化剂或铜催化剂如氯化铜(ii)、硫酸铜的催化剂的情况下,在约25℃至约150℃进行化学式(vi)的化合物与化学式(viia)的化合物的反应。在本发明的具体实施方式中,化学式(viia)或(viic)的化合物,其中r6选自单羧酸基或二羧酸基、酰基氯基、酯基、腈基、醛基、醇基、酰胺基、烷基、烯基、炔基、卤代烷基或酮基,当r6不是单羧酸时,其可以最终被转化为单羧酸。当r6不是单羧酸时,可以通过任何常规方法进行化学式(xviii)的化合物向化学式(viii)的化合物的转化。在溶剂中,用羟基脱保护剂进行化学式(viii)的化合物的脱保护以获得化学式(i)的取代的吡唑化合物。优选地,所述羟基脱保护剂是(例如)碳负载钯。在存在氢气压力的情况下进行脱保护反应。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。优选地,在存在选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行羟基脱保护。更优选地,所述溶剂是甲醇或乙酸乙酯。优选地,在约30℃至约120℃进行用选自无机酸或有机酸或它们的混合物的酸处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的酯的反应。所述无机酸优选地选自盐酸、氢溴酸、硫酸、硝酸和磷酸。更优选地,所述无机酸是盐酸。所述有机酸优选地为乙酸。在本发明的一个具体方面,在存在任选地与酸催化剂结合的缩合剂的情况下,在溶剂中进行化学式(xix)的化合物与化学式(xx)的化合物的缩合反应以获得化学式(xiiia)的化合物。所述缩合剂优选地选自甲醛、多聚甲醛、1,3,5-三噁烷或它们的混合物。更优选地,所述缩合剂是多聚甲醛。所述酸催化剂优选地选自盐酸、硫酸、p-甲苯磺酸、氯化锌或氯化锡(ii)。所述溶剂优选地选自乙酸、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈、n-甲基吡咯烷酮、环己烷、甲苯、二甲苯或苯。更优选地,所述溶剂是乙酸、环己烷、甲苯或二甲苯。优选地,在约30℃至约150℃进行缩合反应。更优选地,所述缩合反应在约40℃至约140℃进行。在存在缩合剂的情况下,通过将化学式(xix)的化合物、化学式(xx)的化合物和缩合剂一起一锅混合,或者通过将化学式(xix)的化合物与缩合剂混合,然后将所得产物与化学式(xx)的化合物反应来进行化学式(xix)的化合物与化学式(xx)的化合物的缩合反应以提供化学式(xiiia)的化合物或者化学式(i)的化合物。优选地,通过将化学式(xix)的化合物与缩合剂混合,然后将所得产物与化学式(xx)的化合物反应来进行所述缩合反应以提供化学式(xiiia)的化合物或者化学式(i)的化合物。在溶剂中,用羟基脱保护剂实施化学式(viiia)的化合物(其中pg1是苄基)的羟基脱保护以获得化学式(i)的取代的吡唑化合物。优选地,所述羟基脱保护剂是(例如)碳负载钯。优选地,在存在氢气压力的情况下进行脱保护反应。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。优选地,在存在选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行羟基脱保护。更优选地,所述溶剂是甲醇或乙酸乙酯。通过在存在碱的情况下的酯水解进行化学式(viiia)的化合物(其中r4是c1-10烷基)的水解以获得化学式(i)的取代的吡唑化合物。优选地,通过在溶剂中,将化学式(viiia)的化合物(其中r4是c1-10烷基)与选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱反应来进行酯水解以获得化学式(i)的取代的吡唑化合物。更优选地,所述碱是氢氧化钠。优选地,在溶剂,如水、四氢呋喃或它们的混合物中进行反应。通过在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱的情况下,在溶剂中使化学式(xxi)的化合物与化学式(vii)的化合物缩合来制备化学式(xix)的化合物以获得化学式(xix)的化合物。更优选地,所述碱是碳酸铯。优选地,在溶剂,如n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮中进行反应。更优选地,所述溶剂是n,n-二甲基甲酰胺。在存在选自季铵盐、鏻盐、冠醚、聚乙二醇和它们的组合的相转移催化剂的情况下进行化学式(xxi)的化合物与化学式(vii)的化合物的缩合反应。优选地,在存在季铵盐或冠醚的情况下进行反应。更优选地,在存在冠醚的情况下进行反应。在本发明的另一个具体方面,在存在任选地与酸催化剂结合的缩合剂的情况下,在溶剂中进行化学式(xx)的化合物与化学式(xxii)的化合物的缩合反应以获得化学式(vib)的化合物。所述缩合剂优选地选自甲醛、多聚甲醛、1,3,5-三噁烷或它们的混合物。更优选地,所述缩合剂是多聚甲醛。所述酸催化剂优选地选自盐酸、硫酸、p-甲苯磺酸、氯化锌或氯化锡(ii)。所述溶剂优选地选自乙酸、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈、n-甲基吡咯烷酮、环己烷、甲苯、二甲苯或苯。更优选地,所述溶剂是乙酸、环己烷、甲苯或二甲苯。优选地,在约30℃至约150℃进行缩合反应。更优选地,所述缩合反应在约40℃至约140℃进行。通过在存在酸催化剂的情况下在溶剂中用肼处理化学式(vib)的化合物来进行化学式(vib)的化合物的环化以获得化学式(vic)的化合物。所述酸催化剂优选地选自乙酸或三氟乙酸。更优选地,所述酸催化剂是乙酸。优选地,在溶剂中在约20℃至约90℃进行化学式(vib)的化合物与肼的环化。更优选地,所述反应在约60℃至约85℃进行。优选地,在存在选自甲醇、乙醇、正丙醇、异丙醇、丁醇、2-甲氧乙醇和2-乙氧乙醇的溶剂的情况下进行化学式(vib)的化合物与肼的反应。更优选地,所述溶剂是异丙醇。然后,在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱的情况下,在溶剂中使化学式(vic)的化合物与化学式(vii)的化合物反应以获得化学式(i)的取代的吡唑化合物。更优选地,所述碱是碳酸铯。优选地,在存在极性非质子溶剂,如(例如)n,n-二甲基甲酰胺的情况下进行反应。还通过任选地在存在选自氢醌、铁催化剂或铜催化剂如氯化铜(ii)、硫酸铜的催化剂情况下,将化学式(vic)的化合物与丙烯酸乙酯反应来制备化学式(i)的取代的吡唑化合物。优选地,在约30℃至约120℃进行化学式(vic)的化合物与丙烯酸乙酯的反应。更优选地,所述反应在约60℃至约110℃进行。在溶剂中,用羟基脱保护剂实施化学式(viiia)的化合物(其中pg1是甲基)的羟基脱保护以获得化学式(i)的取代的吡唑化合物。优选地,所述羟基脱保护剂是(例如)三溴化硼。优选地,在存在选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、二氯甲烷、氯仿和1,2-二氯乙烷的溶剂的情况下进行羟基脱保护。更优选地,所述溶剂是二氯甲烷或乙酸乙酯。可以在存在任选地与酸催化剂结合的缩合剂的情况下,在溶剂中进行化学式(xx)的化合物与化学式(xxiii)的化合物的缩合反应以获得化学式(xxiv)的化合物。所述缩合剂优选地选自甲醛、多聚甲醛、1,3,5-三噁烷或它们的混合物。更优选地,所述缩合剂是多聚甲醛。所述酸催化剂优选地选自盐酸、硫酸、p-甲苯磺酸、氯化锌或氯化锡(ii)。所述溶剂优选地选自乙酸、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈、n-甲基吡咯烷酮、环己烷、甲苯、二甲苯或苯。更优选地,所述溶剂是乙酸、环己烷、甲苯或二甲苯。优选地,在约30℃至约150℃进行缩合反应。更优选地,所述缩合反应在约40℃至约140℃进行。可以在溶剂中,用氨基脱保护剂进行化学式(xxiv)的化合物的脱保护以获得化学式(vic)的化合物。在本发明的另一个具体方面,在存在任选地与酸催化剂结合的缩合剂的情况下,在溶剂中进行化学式(xxv)的化合物与化学式(xx)的化合物的缩合反应以获得化学式(xviiia)的化合物。所述缩合剂优选地选自甲醛、多聚甲醛、1,3,5-三噁烷或它们的混合物。更优选地,所述缩合剂是多聚甲醛。所述酸催化剂优选地选自盐酸、硫酸、p-甲苯磺酸、氯化锌或氯化锡(ii)。所述溶剂优选地选自乙酸、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈、n-甲基吡咯烷酮、环己烷、甲苯、二甲苯或苯。更优选地,所述溶剂是乙酸、环己烷、甲苯或二甲苯。优选地,在约30℃至约150℃进行缩合反应。更优选地,所述缩合反应在约40℃至约140℃进行。在本发明的具体实施方式中,化学式(xxv)的化合物,其中r6是可以转化为羧酸的基团。该基团选自二羧酸基、酰基氯基、酯基、腈基、醛基、醇基、酰胺基、烷基、烯基、炔基、卤代烷基或酮基。当r6不是单羧酸时,可以通过任何常规方法进行化学式(xviiia)的化合物向化学式(viiia)的化合物的转化。可以通过在存在选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、碳酸钠、碳酸钾、碳酸铯、碳酸镁、碳酸氢钠和碳酸氢钾的碱的情况下,在溶剂中使化学式(xxi)的化合物与化学式(viia)的化合物缩合来制备化学式(xxv)的化合物以获得化学式(xxv)的化合物。更优选地,所述碱是碳酸铯。优选地,在溶剂,如n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、乙腈和n-甲基吡咯烷酮中进行反应。更优选地,所述溶剂是n,n-二甲基甲酰胺。在本发明的具体实施方式中,通过施用治疗有效量的化学式(i)的取代的吡唑化合物和药物可用的载体和/或赋形剂,通过本发明合成的化学式(i)的取代的吡唑化合物可以用于治疗与不正确的甲状腺激素活性有关的疾病状况,其选自肥胖症、胰岛素抵抗、血脂异常、代谢综合征、ii型糖尿病,在患有甲状腺机能减退、抑郁症、心血管疾病和皮肤病症的老年患者中用于替代疗法。结晶形式-t15-1:通过将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯在水中混悬,然后用碱水溶液处理来产生结晶形式-t15-1。随后,用酸水溶液使反应混合物酸化以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1。优选地,所述碱选自碳酸钠、碳酸钾、碳酸锂、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、氢氧化锂。更优选地,所述碱是氢氧化钠。更优选地,所述酸是盐酸。优选地,在约10℃至约50℃进行用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的方法。更优选地,在约25℃至约35℃进行用酸水溶液酸化的方法。还可以通过在存在碱水溶液的情况下用适合的羟基脱保护剂处理3-(4-{[7-(pg1-氧)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸或其酯(其中pg1是羟基保护基,例如,苄基)来产生结晶形式-t15-1。随后,用酸水溶液使反应混合物酸化以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-1。优选地,所述适合的羟基脱保护剂是(例如)碳负载钯。优选地,所述碱选自碳酸钠、碳酸钾、碳酸锂、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、氢氧化锂。更优选地,所述碱是氢氧化钠。更优选地,所述酸是盐酸。优选地,在存在氢气压力的情况下进行脱保护反应。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。优选地,在约0℃至80℃下进行在存在碱水溶液的情况下用适合的羟基脱保护剂处理3-(4-{[7-(pg1-氧)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸的方法。更优选地,所述方法在约40℃至约60℃进行。优选地,在约10℃至50℃进行用酸水溶液酸化的方法。更优选地,所述方法在约20℃至约40℃进行。优选地,将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的分离的结晶形式-t15-1在乙酸乙酯中制备成浆液并过滤以除去过量水分。通过几种分析技术,包括x射线粉末衍射(xrpd)、差示扫描量热法(dsc)和热重分析(tga)表征结晶形式-t15-1。图1示出了结晶形式-t15-1的x射线粉末衍射谱图。表1列出了对应于结晶形式-t15-1的衍射图的峰归属和它们的相对强度。表12-θ角相对强度(%)2-θ角相对强度(%)2-θ角相对强度(%)3.71.219.216.027.211.36.18.519.734.827.619.47.424.320.245.129.17.110.735.420.75.130.810.611.936.921.430.131.613.012.3100.021.526.132.110.912.766.222.146.332.510.613.73.022.944.833.08.214.49.223.423.133.91.314.927.824.627.835.12.415.97.724.935.636.33.016.414.125.731.036.93.417.19.226.231.118.725.826.916.6图3示出了结晶形式-t15-1的热重图。在30℃至110℃的温度范围之间观察到了约0.5%的重量损失,如通过tga所确定的。图2示出了以10℃/min的变化速率所实施的结晶形式-t15-1的差示扫描量热图,其中在约212℃观察到了吸热峰。结晶形式-t15-2:通过将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯在水中混悬,然后用碱水溶液处理来产生结晶形式-t15-2。随后,在10℃以下用酸水溶液使反应混合物酸化以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-2。优选地,所述碱选自碳酸钠、碳酸钾、碳酸锂、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾或氢氧化锂。更优选地,所述碱是氢氧化钠。更优选地,所述酸是盐酸。优选地,在约0℃至约50℃进行用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的方法。优选地,将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的分离的结晶形式-t15-2在真空盘式干燥器中干燥以除去过量水分。通过几种分析技术,包括x射线粉末衍射(xrpd)、差示扫描量热法(dsc)和热重分析(tga)表征结晶形式-t15-2。图4示出了结晶形式-t15-2的x射线粉末衍射谱图。结晶形式-t15-2的特征还在于在45℃至180℃的温度范围之间所观察到的约1.5%的重量损失,如通过图6中所示的tga曲线所确定的。结晶形式-t15-2的特征还在于在约201℃至约209℃之间所观察到的吸热峰,如通过图5中所示的dsc所确定的。结晶形式-t15-3:通过用酸处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的酯产生结晶形式-t15-3以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-3。所述酸选自无机酸或有机酸或其组合。所述无机酸优选地选自盐酸、氢溴酸、硫酸、硝酸和磷酸。更优选地,所述无机酸是盐酸。所述有机酸优选地为乙酸。优选地,在约30℃至约120℃进行用无机酸、有机酸或它们的混合物处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的酯的反应。还通过将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯在有机溶剂或有机溶剂和水的化合物中混悬,然后用碱水溶液处理来产生结晶形式-t15-3。随后,用酸水溶液使反应混合物酸化以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式t15-3。所述有机溶剂优选地选自酯溶剂、醇溶剂、烃熔剂、卤代烃溶剂、酮溶剂、醚溶剂、极性非质子溶剂或它们的混合物。优选地,所述碱选自碳酸钠、碳酸钾、碳酸锂、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、氢氧化锂。更优选地,所述碱是氢氧化钠。更优选地,所述酸是盐酸。优选地,在约0℃至150℃进行用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸和用酸酸化的两者方法。更优选地,这些方法是在约10℃至约120℃进行的。优选地,将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的分离的结晶形式-t15-3在乙酸乙酯中制备成浆液并过滤以除去过量水分。还可以通过在有机溶剂或有机溶剂和水的混合物中用适合的羟基脱保护剂处理3-(4-{[7-(pg1-氧)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸(其中pg1是羟基保护基,例如,苄基)来产生结晶形式-t15-3。优选地,所述适合的羟基脱保护剂是(例如)碳负载钯。优选地,在存在氢气压力的情况下进行脱保护反应。氢气压力可以在约10磅每平方英寸(psi)至约300psi的范围内。所述有机溶剂优选地选自酯溶剂、醇溶剂、烃熔剂、卤代烃溶剂、酮溶剂、醚溶剂、极性非质子溶剂或它们的混合物。还可以通过将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸与有机溶剂合并并任选地添加抗-溶剂来产生结晶形式-t15-3。所述有机溶剂优选地选自酯溶剂、醇溶剂、卤代烃溶剂、酮溶剂、醚溶剂、极性非质子溶剂或它们的混合物。所述抗-溶剂优选地选自水、烃熔剂或它们的混合物。在约0℃至所述混合物的回流温度进行3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸与有机溶剂的合并和任选地添加抗-溶剂的方法。图7示出了结晶形式-t15-3的x射线粉末衍射谱图。表2列出了对应于结晶形式-t15-3的衍射图的峰归属和它们的相对强度。表2结晶形式-t15-3未显示出任何重量损失,如通过图9中的tga曲线所确定的。结晶形式-t15-3的特征还在于在约212℃所观察到的吸热峰,如通过图8所示的dsc及其热谱图所确定的。在另一个实施方式中,还通过将结晶形式-t15-2加热至200℃并冷却至20℃至40℃来制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式-t15-3。在另一个实施方式中,还通过将结晶形式-t15-1混悬在有机溶剂中,加热并冷却至20℃至40℃来制备3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式-t15-3。优选地,将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式-t15-1在有机溶剂中的混悬液加热至所述有机溶剂的回流温度,然后冷却至20℃至40℃。所述有机溶剂优选地选自酯溶剂、醇溶剂、烃熔剂、卤代烃溶剂、酮溶剂、醚溶剂、极性非质子溶剂或它们的混合物。结晶形式-t15-4:通过用碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯,在10℃以下用酸酸化,分离固体并空气干燥来产生结晶形式-t15-4以提供3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4。优选地,所述碱选自碳酸钠、碳酸钾、碳酸锂、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、氢氧化锂。更优选地,所述碱是氢氧化钠。更优选地,所述酸是盐酸。优选地,在约0℃至约40℃进行用酸或碱水溶液处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的方法。在一个实施方式中,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的结晶形式-t15-4是含水量在约4.5%至约5.5%的水合物,如通过卡尔费歇尔法(karlfischermethod)所测量的。图10示出了结晶形式-t15-4的x射线粉末衍射谱图。结晶形式-t15-4的特征还在于在30℃至90℃的温度范围之间所观察到的约4.6%的重量损失,如通过图12中所示的tga曲线所确定的。结晶形式-t15-4的特征还在于在约75℃至80℃和约210℃所观察到的吸热峰,如通过图11中所示的dsc所确定的。dmso溶剂化物:通过用dmso处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸来产生dmso溶剂化物以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物。图13示出了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的dmso溶剂化物的x射线粉末衍射谱图。3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的dmso溶剂化物的特征在于在20℃至160℃的温度范围之间所观察到的约19%的重量损失,如图15中所示的tga曲线所确定的。dmso溶剂化物的特征还在于在约133℃至约205℃所观察到的吸热峰,如通过图14中所示的dsc所确定的。在一个实施方式中,当将dmso溶剂化物在水中的混悬液搅拌约30分钟至约5小时时,3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的dmso溶剂化物转化为结晶形式-t15-3。二钠盐:通过将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸或其酯在有机溶剂中混悬并用钠离子源处理所述混悬液来产生3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐。所述有机溶剂优选地选自酯溶剂、醇溶剂、烃熔剂、卤代烃溶剂、酮溶剂、醚溶剂、极性非质子溶剂或它们的混合物。优选地,所述钠离子源选自碳酸钠、碳酸氢钠、氢氧化钠、甲醇钠或叔丁醇钠。更优选地,所述钠离子源是氢氧化钠。在约10℃至40℃进行用钠离子源处理3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的方法。更优选地,所述方法是在约25℃至约35℃进行的。图16示出了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的二钠盐的x射线粉末衍射谱图。3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的二钠盐的特征在于分别在0℃至110℃和110℃至150℃的温度范围之间所观察到的约14.7%和约1.9%的重量损失,如通过图18中所示的tga曲线所确定的。图17示出了3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的二钠盐的dsc。无定形形式:通过提供3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸在适合的溶剂中的溶液或混悬液,任选地添加一种或多种药物可用的载体并分离3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的无定形形式或无定形固体分散体来产生3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的无定形形式或无定形固体分散体。不意欲通过单一作用机制限制本发明,本发明所述的方法包括通过施用3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式t15-1、t15-2、t15-3、t15-4或dmso溶剂化物,或二钠盐,或无定形形式,或它们的组合来治疗与不正确的甲状腺激素活性有关的疾病状况,其选自肥胖症、胰岛素抵抗、血脂异常、代谢综合征、ii型糖尿病,患有甲状腺机能减退、抑郁症、心血管疾病和皮肤病症的老年患者中的替代疗法。在本发明的方法的各个实施方式中,在药物组合物中施用3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式t15-1、t15-2、t15-3、t15-4或dmso溶剂化物,或二钠盐,或无定形形式,或它们的组合。本发明的药物组合物包括药物可用的载体和赋形剂以及3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}-丙酸的结晶形式t15-1、t15-2、t15-3、t15-4或dmso溶剂化物,或二钠盐,或无定形形式,或它们的组合以配制用于向受试者正确施用的组合物。提供了以下实施例以显示本发明的具体实施方式及其合成制备;并且不应将其理解为对本发明范围的限制。实施例:实施例-1:7-羟基-6-甲基-茚满-4-甲醛的制备向三氟乙酸(250ml)和乙酸(250ml)的冷却溶液中添加5-甲基-茚满-4-醇(100.0g,0.6748摩尔)和六亚甲基四胺(94.6g,0.6748摩尔)。将反应混合物加热至95℃并搅拌10小时。反应完成后,用水(500ml)使反应混合物淬灭并用氢氧化钠溶液中和。过滤分离的固体,用水清洗(2×100ml)并吸干。在n,n-二甲基甲酰胺和水的混合物中纯化所得固体以获得浅棕色固体状的7-羟基-6-甲基-茚满-4-甲醛。产量:101.7g(85.46%)hplc纯度:99.45%1h-nmr(400mhz,dmso-d6):δ9.86(1h,s),7.42(1h,s),3.12-3.15(2h,t),2.76-2.80(2h,t),2.17(3h,s),1.99-2.07(2h,m)。质量:175.13(m+-1)熔点:158.50℃实施例-2:7-(苄氧基)-6-甲基-茚满-4-甲醛的制备向7-羟基-6-甲基-茚满-4-甲醛(100.0g,0.5675摩尔)在n,n-二甲基甲酰胺(250ml)中的搅拌溶液中添加碳酸钾(156.8g,1.135摩尔)并搅拌30分钟。随后,将碘化钾(9.42g,0.0567摩尔)和苄基氯(71.8ml,0.6242摩尔)加入至反应混合物并在25℃至30℃搅拌12小时。反应完成后,用水(2500ml)使反应混合物淬灭。过滤分离的固体,用水清洗(2×500ml)并吸干。在己烷中纯化所得固体以获得黄白色固体状的7-(苄氧基)-6-甲基-茚满-4-甲醛。产量:127.5g(84.38%)hplc纯度:99.53%1h-nmr(400mhz,dmso-d6):δ9.99(1h,s),7.54(1h,s),7.34-7.47(5h,m),5.07(2h,s),3.15-3.19(2h,t),2.95-2.99(2h,t),2.22(3h,s),2.02-2.09(2h,m)。质量:267.07(m+-1)熔点:44.96℃实施例-3:4-(苄氧基)-7-(氯甲基)-5-甲基-茚满的制备向7-(苄氧基)-6-甲基-茚满-4-甲醛(125.0g,0.4693摩尔)在二氯甲烷(625ml)中的冷却溶液中添加硼氢化钠(18.64g,0.4928摩尔)和乙酸(125ml)并在20℃至25℃搅拌2小时。反应完成后,用水(625ml)使反应混合物淬灭,分离层并用二氯甲烷(125ml)萃取水层。用碳酸氢钠水溶液(625ml)清洗合并的有机层并原位转入下一步。将亚硫酰氯(38.44ml,0.5303摩尔)在20℃至25℃缓慢加入上述有机层并搅拌2小时。反应完成后,在水(500ml)中使反应混合物淬灭,分离层并用二氯甲烷(250ml)萃取水层。用碳酸氢钠水溶液(62.5g在700ml水中)清洗合并的有机层并蒸馏以获得固体。在己烷中纯化所得固体以获得黄白色固体状的4-(苄氧基)-7-(氯甲基)-5-甲基-茚满。产量:116.5g(86.53%)hplc纯度:99.16%1h-nmr(400mhz,dmso-d6):δ7.33-7.47(5h,m),7.05(1h,s),4.91(2h,s),4.68(2h,s),2.88-2.95(4h,m),2.17(3h,s),2.00-2.07(2h,m)。质量:251.09(碎片峰对应于r-ch2+)熔点:78.57℃实施例-4:4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑的制备向乙酰基丙酮(49.36ml,0.4812摩尔)在丙酮(575ml)和n,n-二甲基甲酰胺(5.75ml)中的搅拌溶液中添加碳酸钾(66.5g,0.4812摩尔)并在回流温度下搅拌1小时。在40℃,向该混合物中添加碘化钾(76.55g,0.4611摩尔)和4-(苄氧基)-7-(氯甲基)-5-甲基-茚满(115.0g,0.4009摩尔)在丙酮(575ml)中的溶液并在回流温度搅拌4小时。反应完成后,过滤反应混合物,用丙酮(2×230ml)清洗并蒸馏滤液以获得残余物。将残余物溶于二氯甲烷(460ml)并用盐酸水溶液(575ml)清洗。用二氯甲烷(115ml)萃取水层,用水(575ml)清洗合并的有机层并蒸馏以获得残余物。将残余物溶于异丙醇(387ml),并添加水合肼(23.87ml,0.4865摩尔)在异丙醇(387ml)和乙酸(51.15ml)中的溶液并在回流温度下搅拌11小时。蒸馏反应混合物以获得残余物。将水(1240ml)加入至残余物,过滤分离的固体,用水(465ml)清洗并吸干。在己烷中纯化固体并干燥以获得黄白色固体状的4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑。产量:111.0g(79.8%)hplc纯度:99.39%1h-nmr(400mhz,dmso-d6):δ11.97(1h,s),7.31-7.44(5h,m),6.53(1h,s),4.82(2h,s),3.50(2h,s),2.86-2.89(2h,t),2.73-2.77(2h,t),2.17(3h,s),1.97-2.09(8h,m)。质量:347.16(m++1)熔点:133.75℃实施例-5:乙基3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯的制备向4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑(109.0g,0.3146摩尔)在n,n-二甲基甲酰胺(545ml)中的搅拌溶液中添加碳酸铯(205.0g,0.6292摩尔)并在25℃至30℃搅拌1小时。随后,将乙基-3-溴代丙酸酯(44.1ml,0.3461摩尔)加入至所述反应混合物并在25℃至30℃搅拌10小时。反应完成后,过滤反应混合物并用水(2616ml)淬灭滤液。过滤分离的固体,用水清洗并吸干。在己烷(872ml)中纯化固体并干燥以获得白色固体状的乙基3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)-丙酸酯。产量:90.0g(64.05%)hplc纯度:97.22%1h-nmr(400mhz,dmso-d6):δ7.33-7.44(5h,m),6.50(1h,s),4.82(2h,s),4.12-4.15(2h,t),4.00-4.05(2h,q),3.50(2h,s),2.85-289(2h,t),2.72-2.79(4h,m),2.10(3h,s),2.08(3h,s),1.97-2.00(2h,m),1.91(3h,s),1.13-1.15(3h,t).质量:447.23(m++1)熔点:72.27℃实施例-6:乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯的制备向乙基3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯(45.0g)在乙酸乙酯(675ml)中的搅拌溶液中,在氢化器中添加在水中(4.5ml)的10%的碳负载钯(4.5g)并在氢气压力(200psi)下,在55℃至60℃搅拌12小时。反应完成后,添加二氯甲烷(450ml),通过床过滤反应混合物并蒸馏滤液以获得固体。在乙酸乙酯中纯化所得固体以获得白色固体状的乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯。产量:31.0g(86.32%)hplc纯度:98.70%1h-nmr(400mhz,dmso-d6):δ8.10(1h,s),6.39(1h,s),4.10-4.14(2h,t),3.99-4.05(2h,q),3.42(2h,s),2.72-2.78(4h,m),2.66-2.70(2h,m),2.08(3h,s),2.02(3h,s),1.93-1.96(2h,m),1.89(3h,s),1.11-1.15(3h,t)。质量:357.17(m++1)熔点:158.34℃实施例-7:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的制备在25℃至30℃,向乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯(72.0g,0.2020摩尔)在四氢呋喃(360ml)中的搅拌混悬液中添加氢氧化钠在水中(504ml)的水溶液(16.16g,0.4040摩尔)并搅拌3小时。反应完成后,添加乙酸乙酯(1080ml)并分离层。用盐酸水溶液(72ml盐酸在432ml水中)使水层酸化,过滤分离的固体,用水清洗并吸干。将固体在水中(1440ml)搅拌1小时,过滤,用水清洗,然后用乙酸乙酯清洗并吸干。将固体在乙酸乙酯(720ml)中进一步搅拌,过滤,用乙酸乙酯清洗并干燥以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。产量:56.0g(84.4%)hplc纯度:99.78%1h-nmr(400mhz,dmso-d6):δ12.31(1h,bs),8.09(1h,s),6.40(1h,s),4.07-4.10(2h,t),3.42(2h,s),2.67-2.76(6h,m),2.08(3h,s),2.02(3h,s),1.95-1.98(2h,m),1.93(3h,s)。质量:329.09(m++1)熔点:212.37℃实施例-8:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(结晶形式-t15-3)的制备向乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯(1.0g)在乙酸(5.0ml)和水(5.0ml)中的搅拌混悬液中添加浓盐酸(2.0ml)并在60℃至65℃搅拌8小时。向该混合物中添加浓盐酸(3.0ml)并在60℃至65℃搅拌3小时。反应完成后,将反应混合物冷却至25℃至30℃,添加水(20.0ml)并搅拌30分钟。过滤分离的固体,用水(10ml)清洗并干燥以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。产量:450.0mg(49.0%)实施例-9:4-(苄氧基)-5-甲基-茚满的制备向5-甲基-茚满-4-醇(30.0g,0.2027摩尔)在n,n-二甲基甲酰胺(70ml)中的搅拌溶液中添加碳酸钾(56.0g,0.4054摩尔)并在25℃至30℃搅拌30分钟。随后,将苄基氯(31.0g,0.2432摩尔)和碘化钾(3.37g,0.0203摩尔)加入至反应混合物并在25℃至30℃搅拌2小时。反应完成后,用水(300ml)使反应混合物淬灭并用乙酸乙酯萃取。干燥乙酸乙酯层并蒸馏。使用己烷:乙酸乙酯通过柱色谱法纯化粗产物以作为液体获得4-(苄氧基)-5-甲基-茚满。产量:29.0g(60.0%)hplc纯度:95.35%1h-nmr(400mhz,dmso-d6):δ7.43-7.45(2h,m),7.14-7.39(3h,m),6.93-6.95(1h,d),6.80-6.86(1h,d),4.85(2h,s),2.85-2.89(2h,m),2.77-2.81(2h,m),2.21(3h,s),1.92-2.17(2h,m)。实施例-10:4-(苄氧基)-7-(氯甲基)-5-甲基-茚满的制备向4-(苄氧基)-5-甲基-茚满(5.0g,0.0209摩尔)在乙酸(20ml)中的透明溶液中添加多聚甲醛(1.17g,0.0419摩尔)并在25℃至30℃吹扫氯化氢气体2小时。反应完成后,将水(50ml)加入至反应混合物,过滤分离的固体并吸干以获得白色固体状的4-(苄氧基)-7-(氯甲基)-5-甲基-茚满。产量:5.0g(83.3%)hplc纯度:90.11%1h-nmr(400mhz,dmso-d6):δ7.32-7.46(5h,m),7.04(1h,s),4.87(2h,s),4.67(2h,s),2.87-2.92(4h,m),2.16(3h,s),1.99-2.16(2h,m)。熔点:70.96℃质量:250.92(碎片峰对应于r-ch2+)实施例-11:3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸的制备向乙基3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯(8.5g,0.019摩尔)在四氢呋喃(42.5ml)中的搅拌混悬液中添加氢氧化钠水溶液(1.5g,0.038摩尔在水(18ml))并在25℃至30℃搅拌3小时。反应完成后,将水(115.7ml)加入至所述反应混合物并用乙酸乙酯(67ml)清洗。用2n盐酸水溶液(20ml)使水层酸化,过滤分离的固体,用水(85ml)清洗并干燥以获得白色固体状的3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸。产量:7.2g(90.4%)hplc纯度:99.15%1h-nmr(400mhz,dmso-d6):δ12.31(1h,bs),7.31-7.45(5h,m),6.52(1h,s),4.83(2h,s),4.09-4.13(2h,t),3.50(1h,s),2.86-2.90(2h,t),2.69-2.76(4h,m),2.11(3h,s),2.10(3h,s),1.77-2.03(2h,m),1.17(3h,t)。熔点:140.18℃质量:419.14(m++1)实施例-12:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(结晶形式-t15-3)的制备将10%碳负载钯(0.3g,在0.3ml水中)、3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)-丙酸(3.0g,0.0077摩尔)和甲醇(50ml)的混悬液加入至氢化器中并在氢气压力(200psi)下在55℃搅拌8小时。反应完成后,将反应混合物冷却至25℃至30℃,并添加二氯甲烷(100ml)。通过床过滤反应混合物并蒸馏滤液以获得粗产物(2.0g)。将乙酸乙酯(30ml)加入至所述粗产物,搅拌1小时,过滤分离的固体并用乙酸乙酯(15ml)清洗以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。产量:1.86g(78.96%)hplc纯度:97.81%1h-nmr(400mhz,dmso-d6):δ12.31(1h,bs),8.09(1h,s),6.40(1h,s),4.07-4.10(2h,t),3.42(2h,s),2.67-2.76(6h,m),2.08(3h,s),2.02(3h,s),1.93-1.97(2h,m),1.90(3h,s)。熔点:210.2℃质量:329.15(m++1)实施例-13:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(结晶形式-t15-1)的制备将10%碳负载钯(0.3g,在0.3ml水中)和氢氧化钠水溶液(0.57g,0.0143摩尔)在水(70ml)中的混悬液加入至氢化器,然后添加3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸(3.0g,0.0077摩尔)。将反应混合物在氢气压力(200psi)下在55℃搅拌5小时。反应完成后,将反应混合物冷却至25℃至30℃,通过床过滤并用水(40ml)清洗。用50%盐酸水溶液(20ml)使滤液酸化并搅拌15分钟。过滤分离的固体,用水(15ml)清洗并吸干。将所述固体在水(60ml)中混悬,搅拌1小时,过滤并用水(18ml),然后用乙酸乙酯(15ml)清洗。将所述固体在乙酸乙酯(30ml)中进一步混悬,搅拌30分钟,过滤,用乙酸乙酯(15ml)清洗并干燥以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。产量:1.4g(59.4%)hplc纯度:98.79%1h-nmr(400mhz,dmso-d6):δ12.31(1h,bs),8.09(1h,s),6.40(1h,s),4.07-4.11(2h,t),3.42(2h,s),2.67-2.76(6h,m),2.08(3h,s),2.02(3h,s),1.93-1.98(2h,m),1.90(3h,s)。熔点:210.5℃质量:329.12(m++1)实施例-14:3-{[7-(苄氧基)-6-甲基-茚满-4-基]-亚甲基}戊烷-2,4-二酮的制备向7-(苄氧基)-6-甲基-茚满-4-甲醛(20.0g,0.0751摩尔)和乙酰丙酮(37.59g,0.3755摩尔)在甲苯(200ml)中的搅拌溶液中添加哌啶(1.5ml)并回流4小时。在回流期间,用dean-stark设备除去副产物水。此后,添加第二批乙酰丙酮(37.59g,0.3755摩尔),回流3小时并蒸馏以获得残余物。通过柱色谱法纯化所得残余物以获得3-{[7-(苄氧基)-6-甲基-茚满-4-基]-亚甲基}戊烷-2,4-二酮。hplc纯度:96.57%1h-nmr(400mhz,dmso-d6):δ7.56(1h,s),7.32-7.46(5h,m),6.87(1h,s),4.98(2h,s),2.96-3.00(4h,m),2.40(3h,s),2.22(3h,s),2.12(3h,s),2.01-2.10(2h,m)。质量:349.11(m++1)实施例-15:3-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}戊烷-2,4-二酮的制备向3-{[7-(苄氧基)-6-甲基-茚满-4-基]亚甲基}-戊烷-2,4-二酮(1.0g,0.00286摩尔)和锌粉(2.06g,0.0315摩尔)的混悬液中添加乙酸(7.0ml)并在25℃至30℃搅拌2小时。将乙酸乙酯(10ml)加入至反应混合物,通过床过滤并用乙酸乙酯清洗。用水清洗滤液,用硫酸钠干燥并蒸馏以获得粗产物,将其通过柱色谱法纯化以获得3-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}戊烷-2,4-二酮。实施例-16:4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑的制备向3-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-戊烷-2,4-二酮(0.5g,0.00142摩尔)在异丙醇(4.0ml)中的搅拌溶液中添加在异丙醇(5.0ml)中的水合肼(0.11g,0.00214摩尔),将所述混合物加热至80℃至85℃,并且搅拌1小时。随后,添加乙酸(0.5ml)并在80℃至85℃搅拌11小时。此后,将水(15ml)加入至所述反应混合物和用二氯甲烷萃取。用硫酸钠干燥有机层并蒸馏以获得粗产物,将其通过柱色谱法纯化以获得4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑。hplc纯度:93.38%1h-nmr(400mhz,dmso-d6):δ11.94(1h,s),7.31-7.44(5h,m),6.53(1h,s),4.82(2h,s),3.50(2h,s),2.86-2.89(2h,t),2.73-2.77(2h,t),2.32(3h,s),1.97-2.09(8h,m)。质量:347.10(m++1)熔点:133.75℃实施例-17:2-羟基-3-甲基苯甲醛的制备向2-甲基酚(100.0g,0.9248摩尔)在乙腈(1000ml)中的搅拌溶液中添加氯化镁(176.1g,1.8496摩尔)、三乙胺(562.0g,5.5488摩尔)和多聚甲醛(166.6g,5.5488摩尔)并在回流温度搅拌2小时。反应完成后,冷却反应混合物并用盐酸在水(400ml)中的水溶液(600ml)淬灭。通过床过滤反应混合物并用乙酸乙酯(2×1000ml)萃取滤液。用饱和氯化钠水溶液(1000ml)清洗合并的有机层并蒸馏以获得粘稠液体状的2-羟基-3-甲基苯甲醛。产量:120.0g(96%)hplc纯度:92.16%1h-nmr(400mhz,dmso-d6):δ11.05(1h,s),10.03(1h,s),7.59-7.60(1h,d),7.47-7.49(1h,d),6.96-7.00(1h,m),2.18(3h,s)。质量:135(m+-1)实施例-18:2-甲酰基-6-甲基苯基-4-甲苯-磺酸酯的制备向2-羟基-3-甲基苯甲醛(110.0g,0.8079摩尔)在n,n-二甲基甲酰胺(330ml)的搅拌溶液中添加碳酸钾(133.8g,0.9681摩尔)并在25℃至30℃搅拌30分钟。将甲苯磺酰氯(169.4g,0.8887摩尔)加入至所述反应混合物并在25℃至30℃搅拌3小时。反应完成后,用水(1650ml)使反应混合物淬灭。过滤分离的固体,用水(2×275ml)清洗并真空干燥以获得黄白色固体状的2-甲酰基-6-甲基苯基-4-甲苯-磺酸酯。产量:198.0g(84.5%)hplc纯度:94.80%1h-nmr(400mhz,dmso-d6):δ9.88(1h,s),7.78-7.81(2h,d),7.64-7.74(2h,m),7.52-7.54(2h,d),7.42-7.46(1h,d),2.42(3h,s),2.04(3h,s)。质量:291(m++1)熔点:60.7℃实施例-19:3-(3-甲基-2-{[(4-甲基苯基)磺酰基]氧}-苯基)丙酸的制备在25℃至30℃,向甲酸(39.64g,0.8611摩尔)和n,n-二甲基甲酰胺(300ml)的搅拌溶液中缓慢加入三甲胺(34.85g,0.3444摩尔)并搅拌30分钟。将2,2-二甲基-1,3-二噁烷-4,6-二酮(米氏酸)(52.13g,0.3617摩尔)和2-甲酰基-6-甲基苯基4-甲苯磺酸酯(100.0g,0.3444摩尔)加入至反应混合物并在100±3℃搅拌3小时。反应完成后,将反应混合物冷却至25℃至30℃,并用水(1200ml)淬灭。过滤分离的固体,用水(2×250ml)清洗并干燥以获得黄白色固体状的3-(3-甲基-2-{[(4-甲基苯基)磺酰基]氧}苯基)丙酸。产量:106.0g(92.17%)hplc纯度:91.26%1h-nmr(400mhz,dmso-d6):δ7.86-7.88(2h,d),7.51-7.53(2h,d),7.15-7.18(3h,m),2.68-2.72(2h,t),2.58(3h,s),2.39-2.43(2h,t),2.04(3h,s)。质量:333.07(m+-1)熔点:106.05℃实施例-20:5-甲基-1-氧-茚满-4-基-4-甲苯-磺酸酯的制备向3-(3-甲基-2-{[(4-甲基苯基)磺酰基]氧}-苯基)丙酸(100.0g,0.2990摩尔)在二氯甲烷(600ml)中的搅拌溶液中缓慢添加亚硫酰氯(53.37g,0.4485摩尔)并在25℃至30℃搅拌1小时。在25℃至30℃,将反应混合物缓慢加入氯化铝(119.63g,0.8971摩尔)在二氯甲烷(600ml)中的混悬液中并搅拌1小时。反应完成后,用1n盐酸水溶液(1000ml)使反应混合物淬灭并分离层。用水(1000ml)清洗有机层,干燥并蒸馏以获得粗产物,将其在甲醇(200ml)中纯化以获得黄白色固体状的5-甲基-1-氧-茚满-4-基-4-甲基苯磺酸酯。产量:70.0g(74.5%)hplc纯度:99.61%1h-nmr(400mhz,dmso-d6):δ7.90-7.92(2h,d),7.52-7.56(3h,m),7.38-7.40(1h,d),2.88-2.91(2h,t),2.58-2.61(2h,t),2.50(3h,s),2.12(3h,s)。质量:316.99(m++1)熔点:126.85℃实施例-21:4-羟基-5-甲基-茚满-1-酮的制备在25℃至30℃,向5-甲基-1-氧-茚满-4-基-4-苯磺酸甲酯(69.0g,0.2183摩尔)在甲醇(345ml)中的搅拌混悬液中添加氢氧化钠(26.2g,0.655摩尔)在水(690ml)中的水溶液。将反应混合物加热至70℃并搅拌5小时。反应完成后,蒸馏反应混合物以除去甲醇,然后用稀盐酸水溶液酸化。过滤分离的固体,用水(200ml)清洗并干燥以获得黄白色固体状的4-羟基-5-甲基-茚满-1-酮。产量:26.0g(73.9%)hplc纯度:99.14%1h-nmr(400mhz,dmso-d6):δ9.20(1h,s),7.14-7.16(1h,d),7.03-7.05(1h,d),2.94-2.97(2h,t),2.58-2.59(2h,t),2.24(3h,s).质量:161.08(m+-1)熔点:210.83℃实施例-22:5-甲基-茚满-4-醇的制备将4-羟基-5-甲基-茚满-1-酮(30.0g)在甲醇(300ml)中的混悬液加入至氢化器,然后添加10%的在水(3.0ml)中的碳负载钯(3.0g)和乙酸(3.0ml)。将反应混合物在氢气压力(200psi)下在55℃搅拌12小时。反应完成后,通过床过滤反应混合物并蒸馏滤液以获得残余物。将所得残余物溶于乙酸乙酯(200ml),用饱和碳酸氢钠水溶液(200ml),然后用水(200ml)清洗。蒸馏有机层以获得黄白色固体状的5-甲基-茚满-4-醇。产量:26.0g(95.2%)hplc纯度:98.31%1h-nmr(400mhz,dmso-d6):δ8.35(1h,s),6.80-6.82(1h,d),6.57-6.58(1h,d),2.74-2.78(4h,m),2.10(3h,s),1.92-1.97(2h,m)。质量:148.93(m++1)熔点:95.79℃实施例-23:5-甲基-茚满-4-基4-甲苯-1-磺酸酯的制备在0℃至-5℃,向5-甲基-1-氧-茚满-4-基4-甲苯-1-磺酸酯(10.0g,0.031摩尔)在二氯甲烷(100ml)中的搅拌溶液中添加三乙基硅烷(40.36ml,0.252摩尔)和三氟化硼醚化物(23.98ml,0.1940摩尔)并在25℃至30℃搅拌12小时。反应完成后,添加水(100ml)并用碳酸钠中和。用乙酸乙酯(2×100ml)萃取反应混合物,干燥合并的有机层并蒸馏以提供粗产物。在甲醇中纯化所述粗产物以获得固体状的5-甲基-茚满-4-基4-甲苯-1-磺酸酯。产量:4.1g实施例-24:5-甲基-茚满-4-基4-甲苯-1-磺酸酯的制备向5-甲基-1-氧-茚满-4-基4-甲苯-1-磺酸酯(15.0g,0.0474摩尔)在二氯甲烷(180ml)中的搅拌溶液中添加水氯化铝(18.96g,0.1422摩尔)和三乙基硅烷(30.27ml,0.1896摩尔)并在25℃至30℃搅拌4小时。反应完成后,添加1n盐酸水溶液(150ml),搅拌30分钟,并分离层。用二氯甲烷(150ml)萃取水层,并用水(2×150ml)清洗合并的有机层并蒸馏以提供粗产物。在甲醇(50ml)中纯化所述粗产物以获得固体状的5-甲基-茚满-4-基4-甲苯-1-磺酸酯。产量:3.37g实施例-25:5-甲基-1h-茚满-4-醇的制备向5-甲基-茚满-4-基4-甲苯-1-磺酸酯(4.0g,0.0126摩尔)在甲醇(20ml)中的搅拌混悬液中添加氢氧化钠(2.02g,0.05摩尔)并在50-55℃搅拌6小时。反应完成后,添加水(20ml)并用稀盐酸酸化。过滤沉淀固体,用水清洗并干燥以获得5-甲基-1h-茚满-4-醇。产量:1.4g实施例-26:乙基3-(3,5-二甲基-1h-吡唑-1-基)-丙酸酯的制备在25℃至30℃,向3,5-二甲基-1h-吡唑(20.0g,0.2080摩尔)和碳酸铯(135.5g,0.4158摩尔)在n,n-二甲基甲酰胺(100ml)中的搅拌混悬液中添加乙基-3-溴代丙酸酯(37.66g,0.2080摩尔)。将反应混合物加热至40℃并搅拌3小时。将反应混合物进一步加热至90℃并搅拌3小时。随后,加入乙基-3-溴代丙酸酯(9.2g,0.050摩尔)并在90℃搅拌3小时。此后,添加水(200ml)并用乙酸乙酯(2×200ml)萃取反应混合物。用水清洗合并的有机层并蒸馏以提供乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯。产量:32.6g(79.9%)hplc纯度:86.14%1h-nmr(400mhz,dmso-d6):δ5.73(1h,s),4.09-4.13(2h,t),4.00-4.06(2h,q),2.76-2.79(2h,t),2.20(3h,s),2.06(3h,s),1.15-1.18(3h,t)。质量:197.09(m++1)实施例-27:乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯的制备向5-甲基-茚满-4-醇(5.0g,0.033摩尔)和多聚甲醛(1.01g,0.033摩尔)在乙酸(50ml)中的搅拌混悬液中添加乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(6.62g,0.033摩尔)并在65℃至68℃搅拌6小时。随后,添加多聚甲醛(0.30g,0.009摩尔)和乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(1.98g,0.01摩尔)并在65℃至68℃搅拌6小时。将反应混合物冷却至25℃至30℃并添加水(150ml)。过滤沉淀固体,用水清洗并吸干。将过滤的固体在乙酸乙酯(70ml)中混悬并在65℃搅拌30分钟。过滤混合物,用乙酸乙酯清洗并干燥以获得白色固体状的乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯。产量:4.45g(37%)hplc纯度:97.34%1h-nmr(400mhz,dmso-d6):δ8.10(1h,s),6.39(1h,s),4.10-4.12(2h,t),3.99-4.05(2h,q),3.42(2h,s),2.66-2.78(6h,m),2.08(3h,s),2.05(3h,s),1.93-1.98(2h,m),1.90(3h,s),1.13-1.17(3h,t)。质量:357.17(m++1)熔点:158.09℃实施例-28:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的制备向乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯(4.0g,0.011摩尔)在四氢呋喃(20ml)中的搅拌溶液中添加氢氧化钠水溶液[氢氧化钠(0.897g)在水(28ml)中]并在25℃至30℃搅拌2小时。将乙酸乙酯(40ml)加入至所述混合物并分离层。用稀盐酸使水层酸化至ph2。过滤沉淀固体,用水(50ml)清洗并吸干。将过滤的固体在乙酸乙酯(40ml)中混悬并在25℃至30℃搅拌30分钟。过滤混合物,用乙酸乙酯清洗并干燥以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。产量:2.2g(59.7%)hplc纯度:99.84%1h-nmr(400mhz,dmso-d6):δ12.31(1h,bs),8.11(1h,s),6.40(1h,s),4.07-4.11(2h,t),3.42(2h,s),2.67-2.76(6h,m),2.08(3h,s),2.02(3h,s),1.93-1.98(2h,m),1.90(3h,s)。质量:329.15(m++1)熔点:212.38℃实施例-29:4-(苄氧基)-5-甲基-茚满的制备向5-甲基-茚满-4-醇(30.0g,0.2027摩尔)在n,n-二甲基甲酰胺(70ml)中的透明溶液中添加碳酸钾(56.0g,0.4054摩尔)并在25℃至30℃搅拌30分钟。随后,添加苄基氯(31.0g,0.2432摩尔)和碘化钾(3.37g,0.0203摩尔)并在25℃至30℃搅拌2小时。反应完成后,添加水(300ml)和乙酸乙酯并分离层。干燥乙酸乙酯层并蒸馏以获得残余物。使用己烷:乙酸乙酯通过柱色谱法纯化所得残余物以作为液体提供4-(苄氧基)-5-甲基-茚满。产量:29.0g(60.0%)hplc纯度:95.35%1h-nmr(400mhz,dmso-d6):δ7.43-7.45(2h,m),7.14-7.39(3h,m),6.93-6.95(1h,d),6.80-6.86(1h,d),4.85(2h,s),2.85-2.89(2h,m),2.77-2.81(2h,m),2.21(3h,s),1.92-2.17(2h,m)。实施例-30:3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯的制备向4-(苄氧基)-5-甲基-茚满(1.0g,0.004摩尔)和多聚甲醛(126mg,0.004摩尔)在乙酸(10ml)中的搅拌混悬液中添加乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(824mg,0.004摩尔)并在85℃至90℃搅拌7小时。随后,添加多聚甲醛(126mg,0.004摩尔)和无水氯化锌(100mg)并在85℃至90℃搅拌10小时。反应完成后,添加水(40ml)并过滤沉淀固体。用水清洗过滤的固体并在己烷中纯化以提供黄白色固体状的乙基3-(4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯。hplc纯度:89.49%1h-nmr(400mhz,dmso-d6):δ7.33-7.44(5h,m),6.50(1h,s),4.82(2h,s),4.12-4.15(2h,t),4.00-4.05(2h,q),3.50(2h,s),2.85-2.87(2h,t),2.73-2.79(4h,m),2.10(3h,s),2.09(3h,s),1.98-2.00(2h,m),1.91(3h,s),1.11-1.15(3h,t)。质量:447.11(m++1)熔点:66.53℃实施例-31:4-甲氧基-5-甲基-茚满的制备向5-甲基-茚满-4-醇(10.0g,0.067摩尔)在n,n-二甲基甲酰胺(40ml)中的搅拌溶液中添加碳酸钾(18.65g,0.134摩尔)并在25℃至30℃搅拌30分钟。随后,添加碘甲烷(14.36g,0.101摩尔)并在25℃至30℃搅拌26小时。将碘甲烷(2.0ml)加入至所述反应混合物并在25℃至30℃搅拌7小时。反应完成后,添加水(160ml)和乙酸乙酯(50ml)并分离层。用水清洗乙酸乙酯层并蒸馏以获得粗产物,将其通过纯化以提供4-甲氧基-5-甲基-茚满。产量:6.0g(55.0%)hplc纯度:99.30%1h-nmr(400mhz,dmso-d6):δ6.91-6.93(1h,d),6.82-6.84(1h,d),3.69(3h,s),2.85-2.89(2h,t),2.77-2.81(2h,t),2.16(3h,s),1.95-1.99(2h,m)。实施例-32:3-(4-{[7-(甲氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯的制备向4-甲氧基-5-甲基-茚满(3.0g,0.018摩尔)和乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(3.63g,0.018摩尔)在乙酸(15ml)中的搅拌溶液中添加多聚甲醛(0.55g,0.018摩尔)和无水氯化锡(ii)(0.3g)并在80℃至85℃搅拌20小时。反应完成后,添加水(100ml)并过滤沉淀的固体。用水清洗过滤的固体并吸干。将所述固体在水(50ml)中混悬并在25℃至30℃搅拌30分钟。过滤混合物,用水清洗并吸干。将所述固体在己烷(30ml)中进一步混悬并在25℃至30℃搅拌1小时。过滤混合物,用己烷(2×10ml)清洗并干燥以获得乙基3-(4-{[7-(甲氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯。产量:2.68g(39.18%)hplc纯度:94.21%1h-nmr(400mhz,dmso-d6):δ6.47(1h,s),4.11-4.15(2h,t),3.99-4.05(2h,q),3.63(3h,s),3.48(2h,s),2.70-2.79(6h,m),2.06(3h,s),2.02(3h,s),1.95-2.00(2h,m),1.89(3h),1.15-1.18(3h,t)。质量:371.21(m++1)熔点:99.93℃实施例-33:3-(4-{[7-(甲氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸的制备向乙基3-(4-{[7-(甲氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸酯(2.5g,0.006摩尔)在四氢呋喃(12.5ml)中的搅拌溶液中添加氢氧化钠水溶液[氢氧化钠(0.539g)在水(12.5ml)中]并在25℃至30℃搅拌2.5小时。反应完成后,添加水(20ml)并用乙酸乙酯(2×20ml)清洗反应混合物。用稀盐酸使水层酸化并在25℃至30℃搅拌30分钟。过滤沉淀物固体,用水(2×20ml)清洗并干燥以获得白色固体状的3-(4-{[7-(甲氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸。产量:1.88g(81.38%)hplc纯度:95.93%1h-nmr(400mhz,dmso-d6):δ12.3(1h,s),6.48(1h,s),4.08-4.12(2h,t),3.64(3h,s),3.48(2h,s),2.84-2.87(2h,t),2.68-2.75(4h,m),2.26(6h,s),2.10-2.23(2h,m),2.08(3h)。质量:343.25(m++1)熔点:158.86℃实施例-34:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的制备在3±3℃,向3-(4-{[7-(甲氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑-1-基)丙酸(1.5g,0.0043摩尔)在二氯甲烷(15ml)中的搅拌混悬液中缓慢加入在二氯甲烷(7.5ml)中的三溴化硼(2.2g,0.0087摩尔)并在25℃至30℃搅拌2小时。随后,添加水(70ml)并搅拌1小时。过滤沉淀的固体,用水(2×20ml)和乙酸乙酯(10ml)清洗并干燥以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。hplc纯度:92.37%1h-nmr(400mhz,dmso-d6):δ6.43(1h,s),4.21-4.24(2h,t),3.50(2h,s),2.70-2.78(4h,m),2.67-2.68(2h,t),2.02(3h,s),2.03(6h,s),1.93-1.97(2h,m)。质量:329.21(m++1)熔点:214.59℃实施例-35:3-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}戊烷-2,4-二酮的制备向4-(苄氧基)-5-甲基-茚满(5.0g,0.0209摩尔)和乙酰丙酮(2.1g,0.0209摩尔)在乙酸(50ml)中的搅拌溶液中添加多聚甲醛(0.63g,0.0209摩尔)和无水氯化锌(0.5g)并在80℃至85℃搅拌18小时。反应完成后,添加水(100ml)并用乙酸乙酯(2×70ml)萃取。用水(2×100ml)清洗合并的乙酸乙酯层,用硫酸钠干燥并蒸馏以提供粗产物,将其通过纯化以提供黄色液体状的3-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}戊烷-2,4-二酮。实施例-36:4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑的制备向3-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-戊烷-2,4-二酮(0.6g,0.0017摩尔)在异丙醇(10ml)中的搅拌溶液中添加水合肼(0.094g,0.0019摩尔)并在80℃至85℃搅拌1小时。随后,添加乙酸(0.6ml)并在80℃至85℃搅拌10小时。反应完成后,添加水(30ml)并用乙酸乙酯(2×20ml)萃取反应混合物。用水(30ml)清洗合并的乙酸乙酯层,用硫酸钠干燥并蒸馏以获得粗产物,将其通过纯化以获得固体状的4-{[7-(苄氧基)-6-甲基-茚满-4-基]甲基}-3,5-二甲基-1h-吡唑。hplc纯度:98.19%1h-nmr(400mhz,dmso-d6):δ7.31-744(5h,m),6.53(1h,s),4.82(2h,s),3.50(2h,s),2.86-2.89(2h,t),2.73-2.77(2h,t),2.09(3h,s),1.97-2.01(8h,m)。质量:347.16(m++1)熔点:132.58℃实施例-37:3-[(7-羟基-6-甲基-茚满-4-基)甲基]-戊烷-2,4-二酮的制备向5-甲基-茚满-4-醇(20.0g,0.1349摩尔)和乙酰丙酮(13.51g,0.1349摩尔)在乙酸(200ml)中的搅拌溶液中添加多聚甲醛(4.05g,0.1349摩尔)并在64℃至68℃搅拌12小时。反应完成后,添加水(200ml)并用二氯甲烷(2×150ml)萃取反应混合物。用水(2×100ml)清洗合并的二氯甲烷层,用硫酸钠干燥并蒸馏以获得粗产物,将其通过纯化以获得浅黄色固体状的3-[(7-羟基-6-甲基-茚满-4-基)甲基]戊烷-2,4-二酮。实施例-38:7-[(3,5-二甲基-1h-吡唑-4-基)甲基]-5-甲基-茚满-4-醇的制备向3-[(7-羟基-6-甲基-茚满-4-基)甲基]戊烷-2,4-二酮(8.0g,0.0307摩尔)在异丙醇(80ml)中的搅拌溶液中添加水合肼(3.078g,0.0615摩尔)并在80℃至85℃搅拌1小时。随后,添加乙酸(8.0ml)并在80℃至85℃搅拌12小时。将反应混合物冷却至25℃至30℃并搅拌2小时。过滤沉淀固体并干燥以获得固体状的7-[(3,5-二甲基-1h-吡唑-4-基)甲基]-5-甲基-茚满-4-醇。hplc纯度:96.74%1h-nmr(400mhz,dmso-d6):δ11.90(1h,s),8.10(1h,s),6.41(1h,s),3.43(2h,s),2.70-2.76(4h,m),2.08(3h,s),1.93-2.02(8h,m)。质量:257.26(m++1)熔点:229.22℃实施例-39:乙基3-{4-[(7-羟基-39-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯的制备将丙烯酸乙酯(4.0ml)和7-[(3,5-二甲基-1h-吡唑-4-基)甲基]-5-甲基-茚满-4-醇(1.0g,0.0039摩尔)的搅拌混合物加热至90℃至95℃并搅拌9小时。添加另一批丙烯酸乙酯(3.0ml)并在90℃至95℃搅拌5小时。用二氯甲烷萃取反应混合物(15ml和10ml)并合并有机层。将己烷缓慢加入合并的有机层并在25℃至30℃搅拌20分钟过滤沉淀固体并干燥以提供白色固体状的乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯。hplc纯度:95.08%1h-nmr(400mhz,dmso-d6):δ8.10(1h,s),6.39(1h,s),4.13-4.17(2h,t),3.99-4.05(2h,q),3.45(2h,s),2.70-2.80(4h,m),2.66-2.68(2h,t),2.14(3h,s),2.10(3h,s),1.95-2.02(2h,m),1.92(3h,s),1.11-1.15(3h,t)。质量:357.30(m++1)熔点:147.09℃实施例-40:3-(3,5-二甲基-1h-吡唑-1-基)-丙酸的制备向乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(4.0g,0.020摩尔)在四氢呋喃(10ml)中的搅拌溶液中添加氢氧化钠水溶液[氢氧化钠(1.63g)在水(10ml)中]并在25℃至30℃搅拌90分钟。通过稀盐酸(30ml)使反应混合物酸化并蒸馏以获得残余物。将甲醇(10ml)加入至残余物并蒸馏以获得粗固体。将二氯甲烷(25ml)和甲醇(25ml)加入至所得粗固体并在25℃至30℃搅拌15分钟。过滤反应混合物以除去氯化钠并蒸馏滤液以获得粗固体。将己烷(25ml)加入至所述粗固体并搅拌30分钟。过滤分离的固体,用己烷(25ml)清洗并干燥以获得白色固体状的3-(3,5-二甲基-1h-吡唑-1-基)丙酸。产量:3.2g(93.3%)1h-nmr(400mhz,dmso-d6):δ6.04(1h,s),4.21-4.24(2h,t),2.81-2.85(2h,t),2.29(3h,s),2.19(3h,s)。质量:169.11(m++1)熔点:260.16℃实施例-41:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的制备向5-甲基-茚满-4-醇(1.5g,0.010摩尔)和多聚甲醛(303mg,0.010摩尔)在乙酸(15ml)中的搅拌混悬液中添加3-(3,5-二甲基-1h-吡唑-1-基)丙酸(1.7g,0.010摩尔)和无水氯化锡(ii)(150mg)并在75℃至80℃搅拌20小时。反应完成后,添加水(100ml),过滤并用水(2×20ml)清洗。在乙酸乙酯中纯化过滤的固体以获得黄白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。hplc纯度:92.93%1h-nmr(400mhz,dmso-d6):δ12.31(1h,bs),8.11(1h,s),6.40(1h,s),4.07-4.11(2h,t),3.42(2h,s),2.67-2.76(6h,m),2.08(3h,s),2.02(3h,s),1.93-1.98(2h,m),1.90(3h,s)。质量:329.28(m++1)熔点:207.06℃实施例-42:乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯的制备向甲基-茚满-4-醇(5.0g,0.033摩尔)在甲苯(50ml)中的搅拌溶液中添加多聚甲醛(1.01g,0.033摩尔)、乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(6.62g,0.033摩尔)和p-甲苯磺酸(5.8g,0.033摩尔)。将混合物加热至110℃至112℃并在110℃至112℃搅拌12小时。在搅拌的同时,使用dean-stark设备除去反应期间形成的副产物水。将反应混合物冷却至25℃至30℃,添加己烷并在25℃至30℃搅拌2小时。过滤沉淀的固体,用己烷(2×15ml)清洗并吸干以获得白色固体状的乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯。hplc纯度:2.5g(94.81%)1h-nmr(400mhz,dmso-d6):δ8.11(1h,s),6.39(1h,s),4.09-4.12(2h,t),3.99-4.05(2h,q),3.43(2h,s),2.66-2.79(6h,m),2.09(3h,s),2.05(3h,s),1.93-1.98(2h,m),1.91(3h,s),1.11-1.15(3h,t)。质量:357.11(m++1)熔点:158.09实施例-43:乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯的制备将3,5-二甲基-1h-吡唑(20.0g,0.2080摩尔)、碳酸钾(57.51g,0.4161摩尔)、18-冠-6(11.0g,0.0416摩尔)在乙酸乙酯(200ml)中的搅拌混悬液加热至55℃至60℃并在55℃至60℃搅拌1小时。随后,在55℃至60℃缓慢加入3-氯丙酸乙酯(36.8ml,0.2705摩尔),然后加热至74℃至79℃并搅拌12小时。添加另一批3-氯丙酸乙酯(2.83ml)并在74℃至79℃搅拌4小时。反应完成后,添加水(200ml)并分离层。用乙酸乙酯(200ml)萃取水层,用水(2×120ml)清洗合并的有机层并蒸馏以获得液体状的乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯。产量:40.0g(98.0%)hplc纯度:97.77%实施例-44:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的制备将乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(5.0g,0.025摩尔)、多聚甲醛(0.687g,0.022摩尔)在乙酸(20ml)中的搅拌混悬液加热至70℃至75℃并搅拌3小时。随后,添加无水氯化锌(500mg)和5-甲基-茚满-4-醇(3.39g,0.022摩尔)在乙酸(5.0ml)中的溶液并在70℃至75℃搅拌8小时。将盐酸水溶液(50ml)加入至反应混合物并在70℃至75℃搅拌4小时。将反应混合物冷却至25℃至30℃,过滤沉淀固体并用水(50ml)清洗。将过滤的固体溶解在n,n-二甲基甲酰胺(25ml)中并在25℃至30℃缓慢加入水(75ml)至透明溶液。过滤沉淀固体,并用水(50ml)清洗。将异丙醇(45ml)加入至湿固体,加热至75℃至80℃并搅拌1小时。将混合物冷却至25℃至30℃并过滤沉淀的固体。用异丙醇(20ml)清洗过滤的固体并干燥以获得白色固体状的3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸。产量:2.0g实施例-45:结晶形式-t15-1的制备在25℃至30℃,向3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(15.0g,0.0456摩尔)在水(150.0ml)中的混悬液中缓慢加入氢氧化钠水溶液(3.65g氢氧化钠在150ml水中)并搅拌10分钟。此后,将反应混合物冷却至26℃至29℃并在26℃至29℃,缓慢加入盐酸水溶液(15ml浓盐酸在90ml水中)并搅拌30分钟。过滤沉淀固体,用水(2×30ml)清洗并吸干。将过滤的固体在水(300ml)中混悬,在25℃至30℃搅拌1小时,过滤并用水清洗。将过滤的固体在乙酸乙酯(150ml)中进一步混悬,在25℃至30℃搅拌15分钟,过滤并用乙酸乙酯(105ml)清洗以获得结晶形式-t15-1。产量:6.5ghplc纯度:98.80%实施例-46:结晶形式-t15-2的制备在25℃至30℃,向3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(15.0g,0.0456摩尔)在水(150ml)中的混悬液中缓慢添加氢氧化钠水溶液(5.48g氢氧化钠在150ml水中)并搅拌10分钟。此后,将反应混合物冷却至0℃至5℃,并在0℃至5℃缓慢加入盐酸水溶液(15ml浓盐酸在90ml水中)并搅拌5分钟。过滤分离的固体,用冷水(2×30ml)清洗并在25℃至30℃在真空盘式干燥器中干燥8小时以获得结晶形式-t15-2。产量:13.0g(86.7%)hplc纯度:99.53%实施例-47:结晶形式-t15-3的制备向乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯(72.0g,0.2020摩尔)在四氢呋喃(360ml)中的混悬液中添加氢氧化钠水溶液(16.16g氢氧化钠在504ml水中)并在25℃至30℃搅拌3小时。反应完成后,添加乙酸乙酯,分离层,并用盐酸水溶液(72ml浓盐酸在432ml水中)使水层酸化。过滤分离的固体,并用水清洗。将过滤的固体在水(1440ml)中混悬,搅拌1小时,过滤并用水清洗,然后用乙酸乙酯清洗。将过滤的固体在乙酸乙酯(720ml)中进一步混悬,搅拌10分钟,过滤并用乙酸乙酯清洗并干燥以获得结晶形式-t15-3。产量:56.0g(84.4%)hplc纯度:99.78%实施例-48:结晶形式-t15-4的制备向3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(5.0g,0.0152摩尔)在水(50ml)中的混悬液中缓慢添加氢氧化钠水溶液(1.83g氢氧化钠在50ml水中)并在25℃至30℃搅拌10分钟。将反应混合物冷却至0℃至5℃,并缓慢加入盐酸水溶液(5ml浓盐酸在30ml水中)并搅拌30分钟。过滤分离的固体,用水(2×30ml)清洗和空气干燥以获得结晶形式-t15-4。产量:3.5g(70.0%)hplc纯度:99.03%实施例-49:dmso溶剂化物的制备将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(5.0g)在二甲基亚砜(dmso)(12ml)中的混悬液在22℃至25℃搅拌5分钟以获得透明溶液。将混合物搅拌5分钟,添加二甲基亚砜(13ml)并在25℃至30℃搅拌2小时。过滤分离的固体,用二甲基亚砜(2×2.5ml)清洗并干燥以获得dmso溶剂化物。产量:4.08g(66.0%)hplc纯度:99.75%实施例-50:结晶形式-t15-3的制备在25℃至30℃将dmso溶剂化物(3.1g)在水(18ml)中混悬并搅拌3小时。过滤混合物,用水(45ml)清洗,并干燥以获得结晶形式-t15-3。产量:1.5g实施例-51:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸二钠盐的制备在20℃至24℃,向3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(5.0g,0.0152摩尔)在丙酮(35ml)中的混悬液中缓慢添加氢氧化钠水溶液(1.34g氢氧化钠在10ml水中),然后添加水(45ml)并在25℃至30℃搅拌30分钟。随后,添加丙酮(100ml)并搅拌1小时。过滤分离的固体,用丙酮(25ml)清洗并干燥以获得所期望的产物。产量:5.0g(88.0%)hplc纯度:99.33%实施例-52:结晶形式-t15-3的制备在25℃至30℃,向3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(1.0g)在n,n-二甲基甲酰胺中的透明溶液中添加水(30ml)。过滤沉淀固体并吸干以提供结晶形式-t15-3。产量:0.9g(90.0%)实施例-53:结晶形式-t15-3的制备将结晶形式-t15-2(350.0mg)在200℃在电炉上加热并冷却至25℃至30℃以获得结晶形式t15-3。实施例-54:结晶形式-t15-3的制备将结晶形式-t15-1(2.0g)在甲醇(30ml)中的混悬液在62℃至65℃搅拌2小时并冷却至25℃至30℃。过滤分离的固体并干燥以获得结晶形式-t15-3。产量:1.38g实施例-55:结晶形式-t15-1的制备将3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(2.0g)在n,n-二甲基甲酰胺(20.0ml)中的透明溶液冷却至-20℃至-25℃,然后在-20℃至-25℃添加甲醇(30ml)和水(30ml)的冷冻混合物。过滤沉淀固体并吸干以提供结晶形式-t15-1。产量:1.60g实施例-56:乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯的制备向3,5-二甲基-1h-吡唑(400.0g,4.161摩尔)和碳酸钾(1150.1g,8.3220摩尔)在乙腈(2l)中的搅拌混悬液中添加18-冠-6乙醚(40g)并将所述混合物加热至72±5℃。随后,将乙基-3-氯代丙酸酯(738.8g,5.4092摩尔)加入至所述反应混合物并在72±5℃搅拌10小时至14小时。反应完成后,将反应混合物冷却至30±5℃,添加水(4800ml)并搅拌20分钟。分离水层并用乙腈(800ml)萃取水层。用20%的氯化钠水溶液(2×2000ml)清洗合并的有机层并馏出以获得乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯。产量:750.0g(91.8%)hplc纯度:99.18%实施例-57:乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯的制备向乙基3-(3,5-二甲基-1h-吡唑-1-基)丙酸酯(30.0g,0.1529摩尔)在乙酸(120ml)中的透明溶液中添加多聚甲醛(4.13g,0.1376摩尔)并在82±3℃搅拌反应混合物2小时。随后,在82±3℃添加在乙酸(120ml)中的5-甲基-茚满-4-醇(18.12g,0.1223摩尔)和浓硫酸(1.5g)并将所述反应混合物在82±3℃搅拌6小时。反应完成后,将混合物冷却至42±3℃并添加水(480ml)。过滤沉淀的固体,用水清洗并干燥以获得白色固体状的乙基3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸酯。产量:35.1g(80.7%)hplc纯度:96.3%实施例-58:3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的无定形形式的制备在28±3℃,向3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸(5.0g)在甲醇(250ml)中的透明溶液中添加聚维酮k-30(5.0g)在甲醇(150ml)中的溶液以获得混合物。将所获得的混合物喷雾干燥以获得3-{4-[(7-羟基-6-甲基-茚满-4-基)甲基]-3,5-二甲基-1h-吡唑-1-基}丙酸的无定形形式。产量:2.5ghplc纯度:98.48%。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1