黎药红葱中两个新颖萘醌二聚体及其制备方法和应用与流程
本发明涉及黎药红葱中两个新颖萘醌二聚体及其制备方法和应用,属于医药生物技术领域。
背景技术:
红葱(eleutherineamericana)为鸢尾科(iridacea)红葱属(eleutherine)植物的全草或鳞茎,又名小红蒜、红葱头、百步还阳。据《中国植物志》记载,鸢尾科红葱属植物全世界约4种,包括e.blulbosa、e.angusta、e.citriodora、e.latifolia,主要分布于东南亚和美洲地区。红葱味苦,性凉,具有清热解毒、利尿除湿、散瘀消肿、止血等功效,其鳞茎或全草均可入药,临床上应用广泛,为我国黎药所收载,主要在我国海南、广西等地区种植栽培。由于来源问题,目前国外对红葱的研究报道几乎空白,而国内对红葱的研究较为肤浅,仅从中得到少量的酚酸、甾体和少量的单倍醌类。在药理活性研究方面,国内对红葱提取物进行筛选发现其具有多方面的药理活性包括抗菌、抗炎、抗病毒、抗氧化、心脑血管作用等。
技术实现要素:
本领域对于红葱新药效成分的研究相对较少,基于此,发明人通过深入研究,从黎药红葱中分离鉴定得到两个新颖的萘醌类二聚体,经鉴定确认其拥有全新的碳结构骨架,具有抗菌,抗肿瘤等药理活性。
本发明采取的技术方案如下:
本发明得到了黎药红葱中两个新颖萘醌二聚体,结构式分别如式1和式2所示:
本发明还具体提供了所述萘醌二聚体的制备方法,包括以下步骤:
s1:取干燥的红葱鳞茎粉末,使用甲醇浸提,甲醇提取液减压浓缩后得到红葱提取物;
s2:将红葱提取物分散于水中,依次使用石油醚、氯仿、乙酸乙酯和正丁醇萃取,得不同极性部位;
s3:取乙酸乙酯部位,进行硅胶柱层析,以石油醚-乙酸乙酯至乙酸乙酯-甲醇为洗脱剂,梯度洗脱,得不同洗脱部位;
s4:取乙酸乙酯-甲醇部位,进行硅胶柱层析,以石油醚-乙酸乙酯为洗脱剂,梯度洗脱,依据薄层层析色谱的rf值合并,得到8个部位fr1.a~h;fr1.a展开剂为体积比1:1的石油醚-乙酸乙酯,rf值为0.25-0.5;fr1.b展开剂为体积比1:8的石油醚-乙酸乙酯,rf值为0.2-0.4;fr1.c展开剂为体积比0:1的石油醚-乙酸乙酯,rf值为0.2-0.5;fr1.d展开剂为体积比99:1的乙酸乙酯-甲醇,rf值为0.3-0.6;fr1.e展开剂为体积比95:5的乙酸乙酯-甲醇,rf值为0.2-0.5;fr1.f展开剂为体积比9:1的乙酸乙酯-甲醇,rf值为0.2-0.5;fr1.g展开剂为体积比6:1的乙酸乙酯-甲醇,rf值为0.3-0.6;fr1.h展开剂为体积比4:1的乙酸乙酯-甲醇,rf值为0.4-0.6。
s5:取fr1.b,使用液相色谱,以甲醇水为洗脱剂,等度洗脱,得到化合物1;
s6:取fr1.d,使用液相色谱,以甲醇水为洗脱剂,等度洗脱,得到化合物2。
优选的,步骤s1中,红葱鳞茎粉末与甲醇的料液比为1kg:(8~10)l。
优选的,步骤s3中,洗脱梯度为:石油醚-乙酸乙酯体积比10:1、5:1、2:1、1:1、0:1,乙酸乙酯-甲醇体积比100:1、80:1、60:1、40:1、30:1、20:1、10:1、0:100。
优选的,步骤s4中,所述乙酸乙酯-甲醇部位为以体积比80:1的乙酸乙酯-甲醇为洗脱剂洗脱所得部位。
优选的,步骤s4中,洗脱梯度为:80:1、60:1、40:1、30:1、20:1、10:1、5:1、0:100。
优选的,步骤s5中,甲醇水的体积分数为55~60%。
优选的,步骤s6中,甲醇水的体积分数为50~55%。
本发明通过抗菌活性筛选和抗肿瘤活性筛选发现,所得化合物1和化合物2具有明显的抗菌和抗肿瘤活性,提示本发明化合物在抗菌、抗肿瘤药物或保健品中具有良好的应用前景。
与现有技术相比,本发明的有益效果是:
a.本发明从红葱中获得两个新颖的萘醌二聚体,实验证明,化合物1和化合物2均能抑制多种细菌的生长,其中对大肠杆菌的抑制效果最为显著,mic值最低,分别为0.06、0.24μg/ml,对耐甲氧西林金黄色葡萄球菌(mrsa)具有显著的抑制活性,mic值分别为0.78、0.32μg/ml。化合物1和化合物2在0.05~0.6μm浓度下对多种肿瘤细胞均有不同程度的杀伤作用,表现出较强的增殖抑制活性,其中,化合物1在0.3μm浓度下,对huh-7、bel-7402、h22、s180的抑制率达到50%以上,在0.6μm浓度下,对bel-7402和s180的抑制率达到90%以上。化合物2在0.3μm浓度下对huh-7、s180和a549的抑制率达到50%以上,在0.6μm浓度下,对huh-7、s180、a549和ags的抑制率达到100%以上。以上结果表明,本发明化合物就有明细的抗菌和抗肿瘤活性,在预防或治疗肿瘤疾病药物或保健品中具有良好的应用前景。
b.本发明化合物的制备方法相对简单,且提取过程中未使用高毒试剂,该方法安全、高效。
附图说明
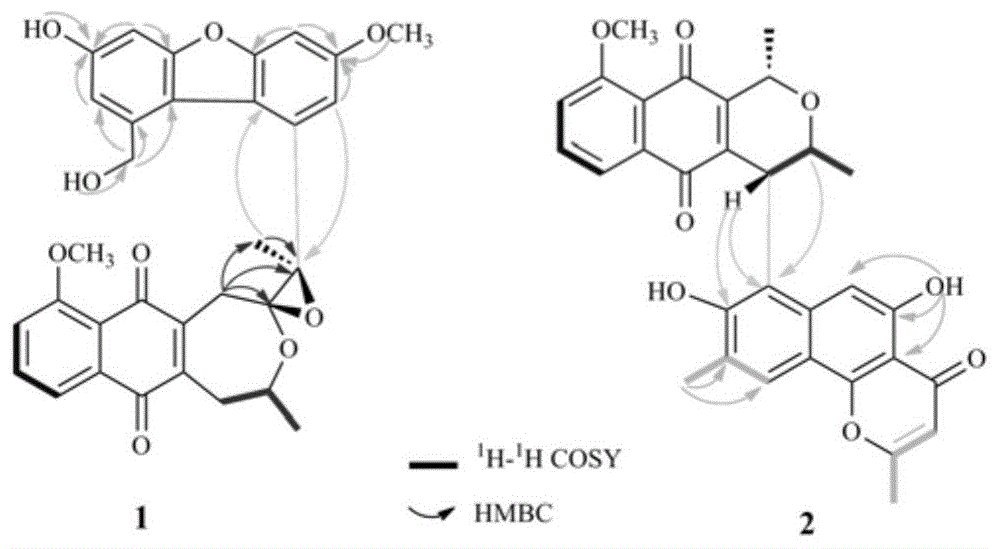
图1化合物1-2的主要cosy和hmbc相关
图2化合物1-2的ecd光谱图
图3化合物1的1h-nmr谱(dmso-d6)
图4化合物1的13c-nmr谱(dmso-d6)
图5化合物1的cosy谱(dmso-d6)
图6化合物1的hsqc谱(dmso-d6)
图7化合物1的hmbc谱(dmso-d6)
图8化合物1的noesy谱(dmso-d6)
图9化合物2的1h-nmr谱(cdcl3)
图10化合物2的13c-nmr谱(cdcl3)
图11化合物2的cosy谱(cdcl3)
图12化合物2的hsqc谱(cdcl3)
图13化合物2的hmbc谱(cdcl3)
图14化合物2的noesy谱(cdcl3)
图15化合物1的hr-esi-ms谱
图16化合物2的hr-esi-ms谱
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。
本发明实施例所用的实验方法如无特殊说明,均为常规方法。
本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1:
萘醌二聚体的制备方法,包括以下步骤:
s1:取干燥的红葱鳞茎粉末,使用甲醇室温浸提3次,料液比1kg:10l,每次提取24h,甲醇提取液减压浓缩后得到红葱提取物;
s2:将红葱提取物分散于水中,依次使用石油醚、氯仿、乙酸乙酯和正丁醇萃取,得不同极性部位;
s3:取乙酸乙酯部位,干法上样,湿法装柱,进行硅胶柱层析(14×100cm,100-200目),以石油醚-乙酸乙酯至乙酸乙酯-甲醇为洗脱剂,梯度洗脱,得不同洗脱部位;洗脱梯度为:石油醚-乙酸乙酯体积比10:1、5:1、2:1、1:1、0:1,乙酸乙酯-甲醇体积比100:1、80:1、60:1、40:1、30:1、20:1、10:1、0:100;
s4:取乙酸乙酯-甲醇部位(80:1),进行硅胶柱层析,以石油醚-乙酸乙酯(80:1、60:1、40:1、30:1、20:1、10:1、5:1、0:100)为洗脱剂,梯度洗脱,依据薄层层析色谱的rf值合并,得到8个部位fr1.a~h;fr1.a展开剂为石油醚-乙酸乙酯(1:1),rf值为0.25-0.5;fr1.b展开剂为石油醚-乙酸乙酯(1:8),rf值为0.2-0.4;fr1.c展开剂为石油醚-乙酸乙酯(0:1),rf值为0.2-0.5;fr1.d展开剂为乙酸乙酯-甲醇(99:1),rf值为0.3-0.6;fr1.e展开剂为乙酸乙酯-甲醇(95:5),rf值为0.2-0.5;fr1.f展开剂为乙酸乙酯-甲醇(9:1),rf值为0.2-0.5;fr1.g展开剂为乙酸乙酯-甲醇(6:1),rf值为0.3-0.6;fr1.h展开剂为乙酸乙酯-甲醇(4:1),rf值为0.4-0.6;
s5:取fr1.b,使用hplc半制备液相色谱,以sb-phenyl色谱柱,以55%甲醇水为洗脱剂,等度洗脱,得到化合物1;
s6:取fr1.d,使用hplc半制备液相色谱,以sb-phenyl色谱柱,以50%甲醇水为洗脱剂,等度洗脱,得到化合物2。
实施例2:
萘醌二聚体的制备方法,包括以下步骤:
s1:取干燥的红葱鳞茎粉末,使用甲醇室温浸提3次,料液比1kg:8l,每次提取24h,甲醇提取液减压浓缩后得到红葱提取物;
s2:将红葱提取物分散于水中,依次使用石油醚、氯仿、乙酸乙酯和正丁醇萃取,得不同极性部位;
s3:取乙酸乙酯部位,干法上样,湿法装柱,进行硅胶柱层析(14×100cm,100-200目),以石油醚-乙酸乙酯至乙酸乙酯-甲醇为洗脱剂,梯度洗脱,得不同洗脱部位;洗脱梯度为:石油醚-乙酸乙酯体积比10:1、5:1、2:1、1:1、0:1,乙酸乙酯-甲醇体积比100:1、80:1、60:1、40:1、30:1、20:1、10:1、0:100;
s4:取乙酸乙酯-甲醇部位(80:1),进行硅胶柱层析,以石油醚-乙酸乙酯(80:1、60:1、40:1、30:1、20:1、10:1、5:1、0:100)为洗脱剂,梯度洗脱,依据薄层层析色谱的rf值合并,得到8个部位fr1.a~h;fr1.a展开剂为石油醚-乙酸乙酯(1:1),rf值为0.25-0.5;fr1.b展开剂为石油醚-乙酸乙酯(1:8),rf值为0.2-0.4;fr1.c展开剂为石油醚-乙酸乙酯(0:1),rf值为0.2-0.5;fr1.d展开剂为乙酸乙酯-甲醇(99:1),rf值为0.3-0.6;fr1.e展开剂为乙酸乙酯-甲醇(95:5),rf值为0.2-0.5;fr1.f展开剂为乙酸乙酯-甲醇(9:1),rf值为0.2-0.5;fr1.g展开剂为乙酸乙酯-甲醇(6:1),rf值为0.3-0.6;fr1.h展开剂为乙酸乙酯-甲醇(4:1),rf值为0.4-0.6;
s5:取fr1.b,使用hplc半制备液相色谱,以sb-phenyl色谱柱,以60%甲醇水为洗脱剂,等度洗脱,得到化合物1;
s6:取fr1.d,使用hplc半制备液相色谱,以sb-phenyl色谱柱,以55%甲醇水为洗脱剂,等度洗脱,得到化合物2。
试验例1:化合物结构鉴定
运用现代结构鉴定技术,确定所得到的活性成分化学结构,发现新颖结构活性成分。结果见图1-图16。
化合物1:
eleucanainonea(1),浅黄色粉末,分子式为c32h28o9(m/z579.1648[m+na]+)(计算值c32h28nao9579.1631)。紫外光谱显示在213,269,308nm处有最大吸收峰。红外光谱显示在νmax3450cm-1,νmax1575,1510,1445cm-1,νmax1215,1175,1065cm-1,νmax1705,1655,1610cm-1有红外吸收。
1hnmr显示有7个芳香质子氢,3个亚甲基氢,1个次甲基氢,2个甲氧基氢及2个甲基氢信号。3个芳香质子氢[h-7(δh7.63,1h,t,j=8.4hz),h-8(δh7.80,1h,t,j=8.4hz),h-9(δh7.55,1h,t,j=8.4hz)]的1h-1h耦合常数表明化合物1存在一个amx结构。同时,4个相邻的质子氢[δh6.68(d,j=2.4hz,h-3′),6.28(d,j=2.4hz,h-5′),7.11(d,j=2.4hz,h-8′),6.31(d,j=2.4hz,h-10′)]表明有2个ab系统结构。13cnmr显示总共32个碳信号,包括2个羰基碳(δc184.0,180.9),15个季碳(δc160.0-71.4),8个次甲基碳(δc118.2,135.2,119.4,111.6,103.8,105.9,99.2),3个亚甲基碳(δc40.3,28.9,63.4),2个甲氧基(δc56.3,55.2),2个甲基碳(δc24.2,20.4)。所有的氢碳信号归属通过hsqc波谱分析得到。综合分析2dnmr波谱数据,说明化合物中含有1a和1b两个结构片段。1a结构与化合物eleutherinf相似,不同之处在于eleutherinf中c-2的甲基在c-2(δc96.2)和c-12(δc71.4)被环醚取代,同时1个甲基(δc24.2)连接在c-12上。这些结构归属被hmbc谱证实。在hmbc光谱图中,h-1(δh3.15,d,j=13.2hz;δh2.29,d,j=13.2hz)和c-2,c-12,c-13相关,ch3-13(δh1.49,s)和c-12相关。
1b结构片段鉴定为4′,13′-dihydroxy-9′-methoxy-6′,7′-benzofuran。在hmbc谱图中,h-3′(δh6.68,d,j=2.4hz)和c-4′(δc158.2)相关,h-5′(δh6.28,d,j=2.4hz)和c-4′,c-6′(δc154.0)相关,h-8′(δh7.11,d,j=2.4hz)和c-7′(δc149.9),c-9′(δc160.0)相关,h-10′(δh6.31,d,j=2.4hz)和c-9′,c-12相关,h-13′(δh4.60,dd,j=12.0,3.0hz;δh4.73,dd,j=12.0,6.6hz)和c-1’(δc114.5),c-2′(δc140.3),c-3′(δc111.6)相关,oh-4′(δh9.74,s)和c-4′相关,oh-13′(δh5.47,dd,j=6.6,3.0hz)和c-13′,och3-9′(δc55.2)和c-9′相关。关键性相关ch3-13和c-12,c-12′相关,h-10′和c-12相关,表明1b结构片段通过一个单键连接在1a结构片段的c-12位。
手性中心的相对构型通过noesy光谱和耦合常数判断。h-4和h-1a,h-1a和ch3-13相关,表明这些氢处于同一平面。c-2和c-12间的醚键处于一个方向。oh-4′和oh-13′,h-3′,h-5′相关,h-10′和och3-9′,h-8′相关,表明它们共平面。由于c-12/c-11′单键,造成了化学扭力,阻碍了该化合物的单晶的形成,因此该化合物的绝对构型不能通过x-ray判断。因此,采用电子圆二色谱法(ecd)测定其绝对构型,判断其绝对构型为2s,4s,12r.
经以上分析,确定化合物1的结构如下所示:
化合物2:
eleucanainoneb(2),红棕色粉末,[m+h]+m/z527.1717(计算值为c31h27o8527.1706)。化合物2在213,250,269处显示最大紫外吸收。红外吸收峰在νmax3425,1710,1665,1615cm-1。1hnmr光谱显示化合物2有一个amx系统[δh7.45(h-6,1h,d,j=8.4hz),δh7.53(h-7,1h,t,j=8.4hz),δh7.22(h-8,1h,d,j=8.4hz)],6个次甲基氢[δh5.14(h-1,q,j=6.6hz),δh4.49(h-3,m),δh4.51(h-4,d,j=5.4hz),δh6.48(h-1′,s),δh7.83(h-5′,s),δh6.26(h-10′,s)],4个甲基氢[δh1.73(1-ch3,d,j=6.6hz),δh1.29(3-ch3,d,j=6.6hz),δh2.96(6′-ch3,s),δh2.48(11′-ch3,s)],1个甲氧基氢[δh3.99(9-och3,s)]及1个羟基氢[δh8.55(s)].13cnmr光谱显示化合物2有31个碳原子,包括4个甲基,1个甲氧基,9个次甲基,14个季碳以及3个羰基信号(δc183.2,183.6,178.9)。碳氢信号归属通过hsqc光谱确定。
详细的1dnmr和2dnmr分析表明,化合物由2a和2b两个部分组成。2a为一个萘醌骨架的化合物,与已知化合物eleutherin结构相似,不同之处在于2a中ch3-1的相对构型为α-位。2b结构片段鉴定为萘酮类骨架,与化合物dihydroeleutherinol结构相似,除了羟基和甲基的位置不同。在hmbc谱图中,oh-2′(δh8.55,s)和c-1′(δc107.5),c-2′,c-3′相关,ch3-6′(δh2.96,s)和c-5′(δc122.7),c-6′(δc136.1)相关.根据c-7′的低场迁移和分子重量,oh-7′连接在c-7′(δc157.3)。h-4和c-7′,c-8′(δc111.5)相关,h-3和c-8′相关,表明2b通过c-8′/c-4单键与2a连接。
化合物2的相对构型通过noesy光谱确定。ch3-3和h-1,h-4相关,表明它们在同一平面。由于c-4/c-8′的单键存在化学扭力,单晶培养不成功。故采用ecd光谱测定化合物2的绝对构型,显示其绝对构型为(1s,3s,4r)-2,与ecd实验光谱一致。
经以上分析,确定化合物2的结构如下所示:
表1.化合物1的nmr数据(dmso-d6)
表2.化合物2的nmr数据(cdcl3)
试验例2:抗菌活性筛选
对实施例1所得的2个化合物进行抗甲氧西林金黄色葡萄球菌、金黄色葡萄球菌、大肠杆菌、铜绿假单胞菌、霍乱弧菌、肺炎杆菌和结核杆菌活性筛选。
菌种分别接种于培养基上,37℃培养24h后,从平板中刮取菌落,以无菌生理盐水稀释,用麦氏比浊管测细菌浓度,配制成9×106cfu/ml细菌稀释液,从各种细菌稀释管中分别取0.05ml菌液,点种于各浓度药物平板及不含药物的对照平板上,37℃培养24h后观察并记录化合物的mic值,实验重复3次,结果见表3。
表3
结果显示:化合物1和化合物2均能抑制多种细菌的生长,其中对大肠杆菌的抑制效果最为显著,mic值最低,分别为0.06、0.24μg/ml。
试验例3:抗肿瘤活性筛选
采用mtt法研究了2种化合物对hepg2、sk-hep-1、huh-7、bel-7402、h22、s180、a549、mcf-7、ags和hccc-9810的增殖抑制作用,用抑制率评价生长抑制效果。结果见表4和表5。
表4化合物1的肿瘤抑制率
表5化合物2的肿瘤抑制率
结果显示:化合物1和化合物2在0.05~0.6μm浓度下对hepg2、sk-hep-1、huh-7、bel-7402、h22、s180、a549、mcf-7、ags和hccc-9810细胞株均有不同程度的杀伤作用,表现出较强的增殖抑制活性。
化合物1在0.3μm浓度下,对huh-7、bel-7402、h22、s180的抑制率达到50%以上,在0.6μm浓度下,对bel-7402和s180的抑制率达到90%以上。
化合物2在0.3μm浓度下对huh-7、s180和a549的抑制率达到50%以上,在0.6μm浓度下,对huh-7、s180、a549和ags的抑制率达到100%以上。
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换。
- 还没有人留言评论。精彩留言会获得点赞!