靶向FAK的化合物及其制备方法和应用与流程
靶向fak的化合物及其制备方法和应用
技术领域
[0001]
本发明涉及化合物技术领域,特别是涉及一种靶向fak的化合物及其制备方法和应用。
背景技术:
[0002]
粘着斑激酶(focal adhesion kinase,fak)是一种非受体型酪氨酸激酶,其在绝大多数种类的肿瘤细胞中都是高度或过度表达的,在肿瘤的发生、发展和转移等各个环节起着至关重要的作用,尤其在肿瘤向恶性侵袭表型演进的过程中起着重要的作用。在理论上阻断fak的表达或者抑制fak的作用,就有可能达到抑制肿瘤细胞侵袭转移发生的目的。因此,fak是一个潜在的肿瘤诊疗靶点。
[0003]
开发对肿瘤高特异性和高灵敏性的放射性药物是必要的。在过去的三十年中,已经设计和发展了一定数量的放射性药物来显像和识别肿瘤组织独特的生物化学特性。fak在肿瘤中的高表达现象,也可以在放射性药物层面上用于肿瘤的早期诊断、治疗和预后评价。
[0004]
现有技术中,普药层面上已经有一些fak小分子抑制剂处于临床研究中,但是现有这些fak小分子抑制剂的肿瘤抑制活性还有待提高。因此有必要研发新的靶向fak的化合物,这些化合物一方面可以直接作为肿瘤生长抑制剂,另一方面也经放射性同位素标记后,可作为靶向fak的肿瘤早期诊断剂。
技术实现要素:
[0005]
本申请的目的在于提供一种靶向fak的化合物及其制备方法和应用。具体包括:
[0006]
本申请第一方面提供了一种靶向fak的化合物,其具有通式(ⅰ)所示的结构:
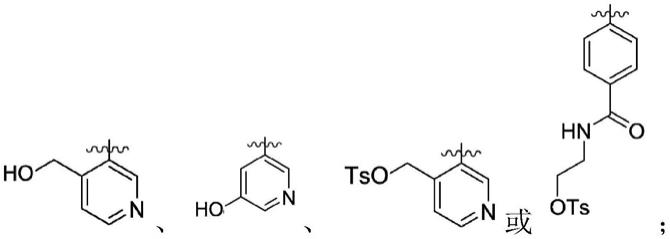
[0007][0008]
其中,r选自
[0009]
r1选自-no2、、
[0010]
r2选自-oh、oh、
[0011]
本申请第二方面提供了本申请第一方面所提供的化合物的制备方法,其包括:
[0012]
1)使式(ⅱ)的化合物:
[0013][0014]
与通式(ⅲ)的化合物:
[0015]
r
’-
nh2(ⅲ),
[0016]
在有机溶剂中,于90-110℃,经对甲苯磺酸催化合成通式(ⅳ)的化合物:
[0017][0018]
其中,r’选自选自
[0019]
r
’1选自-no2、、、、
[0020]
3)以取代化合物的-oh,获得通式(
ⅴ
)或通式(
ⅵ
)的化合物:
[0021]
4)以取代化合物取代化合物中的-no2,获得通式(
ⅶ
)的化合物:
[0022]
[0023]
其中,r
’2选自-oh、
[0024]
5)将通式(ⅳ)-(
ⅶ
)的化合物中的-no2、-oh或-ots中的至少一个,采用含氟化合物取代,获得式(i)的化合物:
[0025][0026]
其中,r如权利要求1所定义;
[0027]
当r为时,不经过含氟化合物取代;
[0028]
当r1为时,还包括以下反应:
[0029][0030]
当r2为时,还包括以下反应:
[0031][0032]
本申请第三方面提供了一种用于制备本申请第一方面所提供的化合物的前体化合物,其选自如下的化合物:
[0033]
[0034][0035]
本申请还提供了本申请第一方面的化合物在制备肿瘤治疗药物和/或肿瘤诊断显像剂中的用途,以及包含本申请第一方面所提供的化合物的药物组合物。
[0036]
本申请所提供的化合物,与粘着斑激酶fak具有较高的亲和性,可作为靶向fak的化合物;进一步地,本申请的靶向fak的化合物对粘着斑激酶fak具有抑制作用,因此可用于制备肿瘤治疗药物;此外,将本申请提供的靶向fak的化合物经过放射性化学标记后,其可以作为肿瘤诊断显像剂,用于制备肿瘤诊断药物。本申请的靶向fak的化合物具有亲和性好、特异性强、选择性高的特点,具有临床应用价值。
附图说明
[0037]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0038]
图1是f-18放射性配体[
18
f]75与其f-19标准品化合物75的hplc共注射分析色谱图。
[0039]
图2是f-18放射性配体[
18
f]83与其f-19标准品化合物83的hplc共注射分析色谱图。
[0040]
图3是f-18放射性配体[
18
f]84与其f-19标准品化合物84的hplc共注射分析色谱图。
[0041]
图4是f-18放射性配体[
18
f]102与其f-19标准品化合物102的hplc共注射分析色谱图。
[0042]
图5是f-18放射性配体[
18
f]103与其f-19标准品化合物103的hplc共注射分析色谱图。
[0043]
图6是小鼠体内化合物[
18
f]75的分布结果。
[0044]
图7是化合物[
18
f]75的摄取阻断实验结果。
具体实施方式
[0045]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0046]
缩写
[0047]
thf
ꢀꢀꢀꢀꢀꢀ
四氢呋喃
[0048]
dmf
ꢀꢀꢀꢀꢀꢀ
n,n-二甲基甲酰胺
[0049]
hatu
ꢀꢀꢀꢀꢀ2--
n,n,n',n'-四甲基脲六氟磷酸酯
[0050]
dipea
ꢀꢀꢀꢀ
n,n-二异丙基乙胺
[0051]
fetos
ꢀꢀꢀꢀ
2-氟乙基甲苯磺酸酯
[0052]-ots
ꢀꢀꢀꢀꢀ
对甲苯磺酰氧基
[0053]
本申请第一方面提供了一种靶向fak的化合物,其具有通式(ⅰ)所示的结构:
[0054][0055]
其中,r选自
[0056]
r1选自-no2、、、、
[0057]
r2选自-oh、
[0058]
在本申请的一些优选的实施方式中,所述靶向fak的化合物,其具有通式(ⅰ)所示的结构:
[0059][0060]
其中,r选自其中,r选自
[0061]
其中,
[0062]
当r2为-oh时,r1选自-no2、、
[0063]
当r2为时,r1选自-no2、、
[0064]
当r2选自时;r1为-no2;
[0065]
当r2为时,r1选自
[0066]
具体地,在本申请第一方面的一些实施方式中,所述靶向fak的化合物选自以下的化合物:
[0067]
[0068]
[0069][0070]
在本申请第一方面的一些实施方式中,当所述化合物中含有氟时,其中至少一个氟被
18
f取代。
[0071]
在本申请第一方面的一些实施方式中,至少一个氟被
18
f取代的化合物选自如下的化合物:
[0072]
[0073][0074]
本申请第二方面提供了本申请第一方面所述的化合物的制备方法,其包括:
[0075]
1)使式(ⅱ)的化合物:
[0076][0077]
与通式(ⅲ)的化合物:
[0078]
r
’-
nh2(ⅲ),
[0079]
在有机溶剂中,于90-110℃,经对甲苯磺酸催化合成通式(ⅳ)的化合物:
[0080][0081]
其中,r’选自选自
[0082]
r
’1选自-no2、、
[0083]
3)以取代化合物的-oh,获得通式(
ⅴ
)或通式(
ⅵ
)的化合物:
[0084][0085]
4)以取代化合物取代化合物中的-no2,获得通式(
ⅶ
)的化合物:
[0086]
[0087]
其中,r
’2选自-oh、
[0088]
5)将通式(ⅳ)-(
ⅶ
)的化合物中的-no2、-oh或-ots中的至少一个,采用含氟化合物取代,获得式(i)的化合物:
[0089][0090]
其中,r如权利要求1所定义;
[0091]
当r为时,不经过含氟化合物取代;
[0092]
当r1为时,还包括以下反应:
[0093][0094]
当r2为时,还包括以下反应:
[0095][0096]
本申请中,式(ⅱ)的化合物可购自商业途径也可通过合成制备,本申请在此不做限定。示例性地,式(ⅱ)的化合物可通过以下方法制备:
[0097]
1)2,4-二硝基苯胺与5-溴-2,4-二氯嘧啶在有机溶剂中、碱性条件下,于60~80℃下反应8-14小时,得到化合物62;
[0098][0099]
2)将步骤1)得到的化合物62中的硝基还原,得到化合物63;
[0100]
3)将步骤2)得到的化合物63中的氨基上连接乙酰基,得到式(ⅱ)的化合物(即化合物65);
[0101][0102]
本申请对式(ⅱ)的化合物的制备中的有机溶剂的选择不做限定,只要能实现本发明的目的即可,例如可以选自dmf或thf。
[0103]
步骤2)中,化合物62中的硝基还原,以及氨基连接乙酰基的方法可采用本领域常用的方法,本申请在此不做限定,例如硝基还原可以通过加氢反应完成,例如可以采用pd/c作为催化剂,与h2反应获得,或者与铁粉氯化铵反应获得;氨基连接乙酰基可通过在碱性条件下,例如在三乙胺溶液中或碳酸钾溶液中,与乙酰氯反应获得。
[0104]
本申请中,采用含氟化合物取代前体化合物中的-no2、-oh或-ots中的至少一个时,所述含氟化合物可以选自:或碱金属氟化物,其中,通常
以与前体化合物中的-oh反应成为以与-no2还原的-nh2反应成为以碱金属氟化物与-ots反应获得-f,所述碱金属氟化物可以选自naf或kf;
[0105]
在本申请的一些实施方式中,当化合物中至少一个氟被
18
f取代时,可采用
18
f标记的化合物与所述前体化合物反应获得,优选地,所述
18
f标记的化合物可以选自或k
18
f;相应地,通常以与前体化合物中的-oh反应成为以与-no2还原的-nh2反应成为以k
18
f与-ots反应获得-18
f。
[0106]
发明人在研究中发现,采用本申请所述的制备方法,反应流程简单;通过先合成前体化合物,然后通过与含氟化合物或者与
18
f标记的含氟化合物反应,即可获得本申请的靶向fak的化合物,或者
18
f标记的靶向fak的化合物,制备方法灵活;此外,采用本申请的反应流程,前体化合物的合成过程中不存在放射性同位素,合成过程更安全;放射性同位素在反应后期加入,减少了放射性同位素的半衰期损失。
[0107]
本申请第三方面提供了一种用于制备本申请第一方面所提供的化合物的前体化合物,其选自如下的化合物:
[0108]
[0109]
[0110][0111]
本申请第四方面提供了本申请第一方面的化合物在在制备肿瘤治疗药物中的用途。发明人在研究中发现,本申请的靶向fak的化合物与fak具有高亲和性,并对fak活性具有一定的抑制作用,进而抑制肿瘤的生长、转移等,因此可用于制备肿瘤治疗药物。
[0112]
本申请第五方面提供了
18
f标记的靶向fak的化合物在制备肿瘤诊断显像剂中的用途。发明人在研究中发现,fak在肿瘤细胞中高表达,通过本申请的
18
f标记的靶向fak的化合物,能够与fak特异性结合,因而能够在肿瘤中富集,结合
18
f标记,可作为肿瘤诊断的显像剂使用,优选地,可用于肿瘤早期诊断中的显像剂。
[0113]
本申请中对肿瘤的种类不做限制,例如,所述肿瘤可以是脑和中枢神经系统(cns)癌症、头颈癌、肾癌、卵巢癌、胰腺癌、肺癌、淋巴瘤、骨髓瘤、肉瘤、乳腺癌和前列腺癌等。示例性脑和中枢cns癌症包括成神经管细胞瘤、少突神经胶质瘤、非典型畸胎样/横纹肌样瘤、脉络丛癌、脉络丛乳头状瘤、室管膜瘤、成胶质细胞瘤、脑膜瘤、神经胶质瘤、少突星形细胞瘤、少突神经胶质瘤和成松果体细胞瘤。示例性卵巢癌包括卵巢透明细胞腺癌、卵巢子宫内膜样腺癌和卵巢浆液性腺癌。示例性胰腺癌包括胰腺导管腺癌和胰腺内分泌肿瘤。示例性肉瘤包括软骨肉瘤、软组织透明细胞肉瘤、尤文肉瘤、胃肠道间质瘤、骨肉瘤、横纹肌肉瘤和未另外指明的(nos)肉瘤。
[0114]
本申请中的肿瘤还可以为罕见病瘤体,例如胸膜间皮瘤、颈静脉球体瘤、神经纤维瘤等。
[0115]
本申请第五方面提供了一种药物组合物,其包含本申请第一方面所提供的的化合物。
[0116]
制备例1.化合物79和化合物80的有机合成
[0117]
合成路线如下:
[0118][0119]
向5-溴-2,4-二氯嘧啶(化合物60)(25.0g,111.1mmol,1equiv)的thf(200ml)溶液中,加入2,4-二硝基苯胺(化合物61)(24.4g,133.3mmol,1.2equiv)的thf(200ml)溶液和碳酸钾(18.4g,133.3mmol,1.2equiv),升温至70℃反应过夜。反应结束后将碳酸钾过滤出来,用乙酸乙酯(50ml*3次)洗涤碳酸钾。收集有机相,旋蒸浓缩滤液,中压制备flash硅胶柱分离(石油醚/乙酸乙酯=10/1至5/1),得到化合物62(21.9g,黄色固体,产率50.8%);
[0120]1h nmr(400mhz,cdcl3,δppm):11.33(s,1h),9.32(d,j=9.2hz,1h),9.21(s,1h),8.59(s,2h).
[0121]
向化合物62(20.0g,53.7mmol,1equiv)的四氢呋喃(100ml)溶液和甲醇(100ml)溶液中,加入铁粉(14.9g,268.5mmol,5equiv)和氯化铵(14.3g,268.5mmol,5equiv)的水(40ml)溶液,升温至80℃反应4h。反应结束后趁热过滤,用甲醇(50ml*3次)洗涤。收集有机相,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物63(15.2g,黑色固体,产率91.2%);
[0122]1h nmr(400mhz,dmso-d6,δppm):8.45(s,1h),8.22(s,1h),6.58(dd,j=3.7hz,5.3hz,1h),5.93(s,1h),5.81(m,1h),4.77(s,2h),4.54(s,2h).
[0123]
13
c nmr(100mhz,dmso-d6,δppm):160.34,158.71,157.45,148.89,145.57,129.19,112.30,103.79,101.00.
[0124]
esi-ms:m/z 313.9800(m+h)
+
[0125]
0℃下向化合物63(15.0g,48.1mmol,1equiv)的无水thf(200ml)溶液中,缓慢加入
乙酰氯(化合物64)(9.2g,120.2mmol,2.5equiv)的无水thf(50ml)稀释后的溶液,0℃下反应5h。反应结束后加水(200ml)猝灭反应,用乙酸乙酯(100ml*3次)萃取。收集有机相,旋蒸浓缩,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至10/1),得到化合物65(12.9g,黄色固体,产率67.8%);
[0126]1h nmr(400mhz,dmso-d6,δppm):10.03(s,1h),9.95(s,1h),8.80(s,1h),8.37(s,1h),7.78(s,1h),7.42(m,2h),2.05(d,j=7.1hz,6h),.
[0127]
13
c nmr(100mhz,dmso-d6,δppm):169.98,168.82,158.75,158.33,158.16,137.88,132.83,127.56,125.52,116.37,114.86,104.22,24.43,23.59.
[0128]
esi-ms:m/z 398.0010(m+h)
+
[0129]
向化合物65(10.0g,25.2mmol,1equiv)的dmf(300ml)溶液中,加入化合物78(4.6g,30.2mmol,1.2equiv)和对甲苯磺酸(1.3g,7.6mmol,0.3equiv),在100℃下搅拌反应5h。反应结束后将反应液倒入200ml水中,然后用乙酸乙酯(50ml*3次)萃取。合并乙酸乙酯层,无水硫酸钠干燥,过滤,旋蒸浓缩滤液,硅胶柱层析分离(石油醚/乙酸乙酯=10/1至5/1),得到化合物79(10.3g,红色固体,产率79.2%);
[0130]1h nmr(400mhz,dmso-d6,δppm):10.37(s,1h),9.99(d,j=2.2hz,2h),9.29(s,1h),8.15(d,j=3.6hz,2h),8.04(s,1h),7.68(m,2h),7.53(d,j=5.8hz,1h),7.30(d,j=6.1hz,1h),6.86(d,j=6.0hz,1h),2.04(d,j=6.6hz,6h).
[0131]
13
c nmr(100mhz,dmso-d6,δppm):170.06,168.78,158.54,157.27,137.31,136.14,132.10,127.65,127.24,119.71,116.69,115.28,114.68,24.54,23.54.
[0132]
esi-hrms m/z calculated for c
20
h
19
brn7o5+516.0626,found 516.0624[m+h]
+
[0133]
向化合物79(500mg,0.97mmol,1equiv)的dmf(6ml)溶液中,加入碳酸钾(200mg,1.45mmol,1.5equiv),加入fetos(423mg,1.94mmol,2equiv)的dmf(5ml)溶液,升温至80℃反应5h。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物80(487mg,红棕色固体,产率89.6%);
[0134]1h nmr(600mhz,dmso-d6,δppm):10.01(s,2h),9.44(s,1h),8.20(s,1h),8.16(s,1h),7.99(s,1h),7.73(d,j=14.8hz,1h),7.66(s,1h),7.49(d,j=12.9hz,1h),7.35(d,j=13.0hz,1h),7.12(d,j=13.7hz,1h),4.74(t,j=2.0hz,1h),4.62(t,j=5.2hz,1h),4.34(t,j=3.9hz,1h),4.26(t,j=3.9hz,1h),2.04(d,j=5.1hz,6h).
[0135]
13
c nmr(100mhz,dmso-d6,δppm):170.03,168.88,162.83,158.34,157.47,145.48,139.86,137.48,134.74,132.43,128.02,125.01,116.84,116.34,115.15,114.79,83.29,81.62,69.73,69.54,24.52,23.58.
[0136]
esi-hrms m/z calculated for c
22
h
22
brfn7o
5+
562.0845,found 562.0848[m+h]
+
[0137]
制备例2.化合物83和化合物84的有机合成
[0138]
合成路线如下:
[0139][0140]
参照制备例1的方法制得化合物79;
[0141]
向化合物79(500mg,0.97mmol,1equiv)的dmf(6ml)溶液中,加入碳酸钾(200mg,1.45mmol,1.5equiv),加入化合物81(911mg,1.94mmol,2equiv)的dmf(5ml)溶液,升温至60℃反应3h。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物82(505mg,黄色固体,产率64.1%);
[0142]1h nmr(600mhz,dmso-d6,δppm):10.02(s,1h),9.99(s,1h),9.43(s,1h),8.20(s,1h),8.16(s,1h),7.97(s,1h),7.76-7.73(m,3h),7.62(s,1h),7.49(d,j=8.2hz,1h),7.39-7.38(m,3h),7.06(d,j=9.5hz,1h),4.30(dd,j=2.0hz,10.6hz,1h),4.16-4.11(m,4h),4.05-4.02(m,1h),2.32(s,3h),2.04(d,j=13.9hz,6h),1.25(d,j=13.4hz,6h).
[0143]
esi-hrms m/z calculated for c
34
h
37
brn7o
10
s
+
814.1501,found 814.1497[m+h]
+
[0144]
向化合物82(100mg,0.12mmol,1equiv)的dmf(5ml)溶液中,加入碳酸钾(2mg,0.01mmol,0.1equiv)、kryptofix 222(cas号23978-09-8,以下简称k 2.2.2)(45mg,0.12mmol,1equiv)和kf(14mg,0.24mmol,2equiv),升温至100℃反应1h。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物83(71mg,黄色固体,产率90.1%);
[0145]1h nmr(600mhz,cd3od,δppm):8.07(s,1h),7.93(s,1h),7.68(d,j=2.0hz,1h),7.59(dd,j=2.1hz,9.1hz,1h),7.51(d,j=8.7hz,1h),7.40(d,j=2.0hz,8.1hz,1h),7.06(d,j=9.1hz,1h),4.31-4.29(m,1h),4.25-4.19(m,3h),3.84-3.81(m,1h),3.76-3.73(m,1h),2.14(s,3h),2.11(s,3h),1.40(s,3h),1.38(s,3h).
[0146]
esi-hrms m/z calculated for c
27
h
30
brfn7o
7+
662.1369,found 662.1359[m+h]
+
[0147]
向化合物83(20mg,0.03mmol,1equiv)的dmf(3ml)溶液中,加入1m的盐酸(200μl),升温至100℃反应20min。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物84(16mg,黄色固体,产率85.6%);
[0148]1h nmr(600mhz,cd3od,δppm):8.09(s,1h),7.87(s,1h),7.65(s,1h),7.56(d,j=9.7hz,1h),7.48(d,j=7.9hz,1h),7.36(d,j=9.2hz,1h),7.09(d,j=8.9hz,1h),4.77-4.44(m,2h),4.21-4.12(m,2h),4.01-3.98(m,2h),2.14(d,j=8.2hz,6h).
[0149]
esi-hrms m/z calculated for c
24
h
26
brfn7o
7+
622.1056,found 622.1060[m+h]
+
[0150]
制备例3.化合物75的有机合成
[0151]
合成路线如下:
[0152][0153]
参照制备例1的方法制得化合物65;
[0154]
向n-乙酰-β-丙氨酸(化合物67)(5.0g,38.1mmol,1equiv)的二氯甲烷(100ml)溶液中,加入hatu(17.3g,45.7mmol,1.2equiv)和dipea(14.7g,114.3mmol,3equiv),室温下搅拌1h。加入2-氨基-4-硝基苯酚(化合物66)(7.0g,45.7mmol,1.2equiv),室温下过夜搅拌。析出固体,过滤,用二氯甲烷(50ml*3)洗涤固体,得到化合物68(8.9g,黄色固体,产率88.0%);
[0155]1h nmr(400mhz,dmso-d6,δppm):9.40(s,1h),8.93(s,1h),7.90(t,j=9.0hz,2h),6.99(d,j=8.6hz,1h),3.30(d,j=5.0hz,2h),2.57(s,2h),1.76(s,3h).
[0156]
13
c nmr(100mhz,dmso-d6,δppm):176.04,174.72,159.54,144.49,132.24,126.04,122.22,120.20,41.70,40.52,28.07.
[0157]
esi-hrms m/z calculated for c
11
h
14
n3o
5+
268.0928,found 268.0930[m+h]
+
.
[0158]
向化合物68(5.0g,18.7mmol,1equiv)的四氢呋喃(50ml)溶液和甲醇(50ml)溶液中,加入铁粉(5.2g,93.5mmol,5equiv)和氯化铵(5.0g,93.5mmol,5equiv)的水(10ml)溶液,升温至80℃反应3h。反应结束后趁热过滤,用甲醇(50ml*3次)洗涤。收集有机相,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物69(3.9g,黑色固体,产率89.2%);
[0159]1h nmr(400mhz,dmso-d6,δppm):9.24(s,1h),8.57(s,1h),7.90(s,1h),6.86(s,1h),6.49(d,j=8.2hz,1h),6.13(d,j=8.1hz,1h),4.49(s,2h),3.20(d,j=5.7hz,2h),2.45(d,j=6.4hz,1h),1.70(s,3h).
[0160]
13
c nmr(100mhz,dmso-d6,δppm):170.29,169.69,141.44,139.21,126.96,117.35,111.41,108.99,36.46,35.77,23.02.
[0161]
esi-hrms m/z calculated for c
11
h
15
n3nao
3+
260.1006,found 260.1003[m+h]
+
.
[0162]
向化合物65(5.0g,12.5mmol,1equiv)的dmf(100ml)溶液中,加入化合物69(4.4g,18.7mmol,1.5equiv)和对甲苯磺酸(0.6g,3.7mmol,0.3equiv),在100℃下搅拌反应5h。反应结束后将反应液倒入200ml水中,然后用乙酸乙酯(50ml*3次)萃取。合并乙酸乙酯层,无水硫酸钠干燥,过滤,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物74(5.5g,白色固体,产率74.7%);
[0163]1h nmr(600mhz,dmso-d6,δppm):10.00(s,1h),9.98(s,1h),9.27(s,1h),9.17(s,1h),8.95(s,1h),8.06(s,1h),8.04(s,1h),7.91(s,1h),7.88(t,j=5.2hz,1h),7.64-7.59(m,2h),7.31(dd,j=1.9hz,8.7hz,1h),7.21(dd,j=3.5hz,9.9hz,1h),6.60(d,j=8.7hz,1h),3.25-3.23(m,2h),2.51(t,j=5.7hz,2h),2.04(s,3h),2.02(s,3h),1.75(s,3h)。
[0164]
esi-hrms m/z calculated for c
25
h
28
brn8o
5+
599.1361,found 599.1359[m+h]
+
.
[0165]
向化合物74(100mg,0.17mmol,1equiv)的dmf(2ml)溶液中,加入碳酸钾(34mg,0.25mmol,1.5equiv)和碘化钠(3mg,0.02mmol,0.1equiv),加入fetos(74mg,0.34mmol,2equiv)的dmf(1ml)溶液,在70℃下过夜反应。过滤,浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物75(61mg,白色固体,产率55.7%);
[0166]1h nmr(600mhz,dmso-d6,δppm):9.98(s,2h),9.06(s,1h),8.85(s,1h),8.08(d,j=9.1hz,2h),7.85(s,2h),7.60-7.58(m,2h),7.37(d,j=8.4hz,1h),7.32(d,j=8.2hz,1h),6.78(d,j=8.8hz,1h),4.75(t,j=3.5hz,1h),4.67(t,j=3.3hz,1h),4.18(t,j=3.6hz,1h),4.13(t,j=3.4hz,1h),3.25(d,j=6.3hz,2h),2.48(m,2h),2.04(s,3h),2.03(s,3h),1.74(s,3h).
[0167]
13
c nmr(100mhz,dmso-d6,δppm):170.09,169.82,168.91,167.85,158.95,157.57,157.22,144.74,137.06,134.49,128.06,127.87,127.64,116.99,116.78,115.68,115.39,113.85,83.47,82.37,69.48,36.80,35.80,24.59,23.63,23.19.
[0168]
esi-hrms m/z calculated for c
27
h
31
brfn8o
5+
645.1579,found 645.1581[m+h]
+
.
[0169]
制备例4.化合物76和化合物77的有机合成
[0170]
合成路线如下:
[0171][0172]
按照制备例1的方法制备得到化合物65。
[0173]
向n-乙酰基乙二胺(化合物71)(5.0g,49.0mmol,1equiv)的二氯甲烷(100ml)溶液中,加入hatu(22.2g,58.8mmol,1.2equiv)和dipea(18.9g,147.0mmol,3equiv),室温下搅拌1h。加入5-硝基水杨酸(化合物70)(10.2g,58.8mmol,1.2equiv),室温下过夜搅拌。析出固体,过滤,用二氯甲烷(50ml*3)洗涤固体,得到化合物72(6.6g,黄色固体,产率91.2%);
[0174]1h nmr(600mhz,dmso-d6,δppm):13.63(s,1h),9.19(s,1h),8.81(d,j=1.9hz,1h),8.23(d,j=9.3hz,1h),7.95(s,1h),7.07(d,j=9.0hz,1h),3.34(dd,j=5.8hz,11.7hz,2h),3.22(dd,j=6.1hz,12.1hz,2h),1.77(s,3h).
[0175]
13
c nmr(100mhz,dmso-d6,δppm):175.08,172.58,171.23,144.28,134.08,130.49,124.02,121.44,43.36,28.10.
[0176]
esi-hrms m/z calculated for c
11
h
14
n3o
5+
268.0928,found 268.0930[m+h]
+
.
[0177]
向化合物72(5.0g,18.7mmol,1equiv)的四氢呋喃(50ml)溶液和甲醇(50ml)溶液中,加入铁粉(5.2g,93.5mmol,5equiv)和氯化铵(5.0g,93.5mmol,5equiv)的水(10ml)溶液,升温至80℃反应3h。反应结束后趁热过滤,用甲醇(50ml*3次)洗涤。收集有机相,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物73(3.8g,黑色固体,产率86.3%);
[0178]1h nmr(400mhz,dmso-d6,δppm):11.34(s,1h),8.66(s,1h),8.02(s,1h),7.01(s,1h),6.68-6.61(m,2h),4.51(s,2h),3.17-3.14(m,4h),1.78(s,3h).
[0179]
13
c nmr(100mhz,dmso-d6,δppm):169.93,169.33,151.13,140.79,121.11,117.88,116.21,112.85,39.28,38.67,23.05.
[0180]
esi-hrms m/z calculated for c
11
h
15
n3nao
3+
260.1006,found 260.1003[m+h]
+
;
[0181]
向化合物65(5.0g,12.5mmol,1equiv)的dmf(100ml)溶液中,加入化合物73(4.4g,18.7mmol,1.5equiv)和对甲苯磺酸(0.6g,3.7mmol,0.3equiv),在100℃下搅拌反应5h。反应结束后将反应液倒入200ml水中,然后用乙酸乙酯(50ml*3次)萃取。合并乙酸乙酯层,无水硫酸钠干燥,过滤,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物76(6.1g,白色固体,产率82.3%);
[0182]1h nmr(600mhz,dmso-d6,δppm):11.89(s,1h),10.01(s,1h),9.97(s,1h),8.95(s,
1h),8.64(d,j=5.4hz,1h),8.08-8.07(m,2h),7.93(t,j=5.5hz,1h),7.75(s,1h),7.64-7.62(m,2h),7.44(dd,j=2.1hz,9.0hz,1h),7.20(d,j=8.0hz,1h),6.69(d,j=8.6hz,1h),3.27-3.26(m,2h),3.17(dd,j=6.0hz,12.1hz,2h),2.04(s,3h),2.01(s,3h),1.75(s,3h).
[0183]
13
c nmr(600mhz,dmso-d6,δppm):170.08,168.98,168.77,159.20,157.58,156.87,155.20,136.80,131.90,131.40,127.66,127.12,121.20,117.20,116.58,115.92,115.40,93.26,38.56,24.52,23.46,23.14.
[0184]
esi-hrms m/z calculated for c
25
h
28
brn8o
5+
599.1361,found 599.1359[m+h]
+
.
[0185]
向化合物76(100mg,0.17mmol,1equiv)的dmf(2ml)溶液中,加入碳酸钾(34mg,0.25mmol,1.5equiv)和碘化钠(3mg,0.02mmol,0.1equiv),加入fetos(74mg,0.34mmol,2equiv)的dmf(1ml)溶液,在70℃下过夜反应。过滤,浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物77(80mg,白色固体,产率73.5%);
[0186]1h nmr(600mhz,dmso-d6,δppm):10.00(s,1h),9.97(s,1h),9.20(s,1h),8.10-8.08(m,3h),7.89(t,j=5.3hz,1h),7.74(s,1h),7.70(d,j=8.7hz,1h),7.60(s,1h),7.57(d,j=8.2hz,1h),7.41(d,j=8.4hz,1h),6.87(d,j=8.5hz,1h),4.81(t,j=3.2hz,1h),4.73(t,j=3.4hz,1h),4.29(t,j=3.2hz,1h),4.24(t,j=3.3hz,1h),3.30-3.29(m,2h),3.16(dd,j=6.0hz,12.0hz,2h),2.04(s,3h),2.03(s,3h),1.76(s,3h).
[0187]
13
c nmr(100mhz,dmso-d6,δppm):170.02,168.87,165.29,158.72,157.51,157.29,150.97,137.12,134.73,132.14,127.42,123.81,123.64,122.09,117.00,115.26,114.20,83.19,82.09,69.16,69.03,38.84,24.50,23.58,23.10.
[0188]
esi-hrms m/z calculated for c
27
h
31
brfn8o
5+
645.1579,found 645.1581[m+h]
+
.
[0189]
制备例5.化合物95的有机合成
[0190]
合成路线如下:
[0191][0192]
参照制备例1的方法制得化合物79。
[0193]
室温下向6-氟烟酸(化合物92)(15.0g,106.3mmol,1equiv)的1,4-二氧六环(500ml)溶液中,加入2,3,5,6-四氟苯酚(化合物86)(17.6g,106.3mmol,1equiv)和二环己基碳二亚胺dcc(24.1g,116.9mmol,1.1equiv),室温下搅拌过夜。反应结束后将副产物dcu过滤出来,滤液旋蒸浓缩,中压制备flash硅胶柱分离(石油醚/乙酸乙酯=20/1至5/1),得
到化合物93(24.3g,白色固体,产率79.2%).
[0194]1h nmr(400mhz,cdcl3,δppm):9.08(s,1h),8.57(m,1h),7.13(m,2h).
[0195]
向化合物79(10.0g,19.4mmol,1equiv)的四氢呋喃(50ml)溶液和甲醇(50ml)溶液中,加入铁粉(5.4g,97.0mmol,5equiv)和氯化铵(5.1g,97.0mmol,5equiv)的水(20ml)溶液,升温至80℃反应2h。反应结束后趁热过滤,用甲醇(50ml*3次)洗涤。收集有机相,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物94(8.4g,白色固体,产率89.2%);
[0196]1h nmr(400mhz,dmso-d6,δppm):9.99(s,2h),8.70(s,1h),8.48(s,1h),8.05(d,j=16.9hz,2h),7.67(d,j=6.8hz,2h),7.39(d,j=8.7hz,1h),6.75(s,1h),6.58(d,j=8.2hz,1h),6.41(d,j=8.3hz,1h),4.22(m,2h),2.07(d,j=8.2hz,6h).
[0197]
13
c nmr(100mhz,dmso-d6,δppm):169.95,168.71,158.93,157.30,156.86,139.60,136.76,136.48,133.01,131.65,127.71,127.36,116.69,115.33,114.51,108.79,107.36,92.38,24.42,23.42.
[0198]
esi-ms:m/z 486.0881(m+h)
+
[0199]
向化合物94(1.0g,2.0mmol,1equiv)的dmso(10ml)溶液中,加入化合物93(1.1g,4.0mmol,2equiv)和dipea(0.2g,1.5mmol,0.75equiv),升温至60℃反应1h。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物95(0.9g,绿色固体,产率75.0%);
[0200]1h nmr(400mhz,dmso-d6,δppm):10.01(s,1h),9.87(s,1h),9.72(s,1h),9.13(s,1h),9.04(s,1h),8.75(s,1h),8.44(t,j=4.8hz,1h),8.09(s,2h),7.63(m,3h),7.30(m,3h),6.67(d,j=5.8hz,1h),2.05(s,3h),1.97(s,3h).
[0201]
13
c nmr(100mhz,dmso-d6,δppm):170.28,168.85,159.09,157.66,157.14,148.63,48.38,145.74,142.51,142.42,136.94,132.82,131.75,129.56,127.86,127.40,125.25,118.86,116.95,116.32,115.60,110.50,110.18,109.86,24.60,23.68.
[0202]
esi-ms:m/z 609.1000(m+h)
+
[0203]
制备例6.化合物102和化合物103的合成
[0204]
合成路线如下:
[0205]
[0206][0207]
参照制备例1的方法制得化合物79。
[0208]
参照制备例5的方法制得化合物93。
[0209]
向化合物79(1.0g,1.9mmol,1equiv)的dmf(50ml)溶液中,加入n,n-二甲基-溴乙酰胺(化合物96)(0.6g,3.8mmol,2equiv)、碳酸钾(0.5g,3.8mmol,2equiv)和碘化钾(33.2mg,0.2mmol,0.1equiv),升温至50℃反应3h。反应结束加水(100ml)析出固体,过滤干燥收集固体,得到化合物97(0.8g,黄色固体,产率72.3%);
[0210]1h nmr(400mhz,dmso-d6,δppm):10.02(s,2h),9.35(s,1h),8.15(s,1h),8.10(s,1h),7.94(s,1h),7.63(d,j=7.7hz,1h),7.57(s,1h),7.46(d,j=8.8hz,1h),7.31(d,j=7.6hz,1h),6.96(d,j=8.9hz,1h),4.87(s,2h),2.89(s,3h),2.73(s,3h),1.99(d,j=5.6hz,6h).
[0211]
13
c nmr(100mhz,dmso-d6,δppm):174.79,168.85,166.96,158.40,157.37,145.79,139.67,137.37,134.39,130.18,124.91,117.00,116.12,115.34,114.88,67.32,36.00,35.51,24.50,23.55.
[0212]
esi-ms:m/z 601.1156(m+h)
+
[0213]
将化合物97(100mg,0.16mmol,1equiv)溶于5ml四氢呋喃和5ml甲醇的混合溶液中,加入铁粉(22.4mg,0.4mmol,2.5equiv)和氯化铵(21.2mg,0.4mmol,2.5equiv)的水(1ml)溶液,升温至80℃反应3h。反应结束后趁热过滤,用甲醇(10ml*3次)洗涤。收集有机相,旋蒸浓缩滤液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物98(80mg,白色固体,产率87.7%);
[0214]1h nmr(400mhz,dmso-d6,δppm):10.00(s,2h),8.83(s,1h),8.07(d,j=6.9hz,2h),7.63(d,j=7.3hz,2h),7.42(d,j=8.6hz,1h),6.80(s,1h),6.66(d,j=8.4hz,1h),6.55(s,1h),4.61(s,2h),4.56(s,2h),2.95(s,3h),2.82(s,3h),2.06(d,j=8.0hz,6h).
[0215]
13
c nmr(100mhz,dmso-d6,δppm):175.03,170.05,168.85,168.46,158.88,
157.41,157.06,141.41,138.54,136.95,135.20,131.93,127.87,127.60,116.90,115.31,114.17,108.42,106.85,68.29,36.12,35.51,25.02,24.50.
[0216]
esi-ms:m/z 571.1415(m+h)
+
[0217]
0℃下向fmoc-2-氨基-5-(叔丁氧基)-5-氧代戊酸(化合物99)(90mg,0.21mmol,1equiv)的无水dmf(3ml)溶液中加入dipea(32.5mg,0.25mmol,1.2equiv)和hatu(79.8mg,0.21mmol,1equiv)的无水dmf(3ml)溶液,0℃下搅拌0.5h。加入化合物98(96.9mg,0.17mmol,0.8equiv)的无水dmf(3ml)溶液,继续搅拌2h。反应结束后加水(50ml)析出固体,固体为粗产物,这里未纯化直接进行下一步反应,得到化合物100粗产物(70mg,白色固体,产率34.1%)。
[0218]
向化合物100(50mg,0.05mmol,1equiv)的四氢呋喃(3ml)溶液中,加入四正丁基氟化铵的1m四氢呋喃溶液(0.1ml,0.1mmol,2equiv)溶液,室温下反应0.5h。反应结束后旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物101(34.3mg,白色固体,产率91.1%);
[0219]1h nmr(600mhz,dmso-d6,δppm):10.46(s,1h),10.29(s,1h),10.20(s,1h),9.08(s,1h),8.18(s,1h),8.11(s,1h),8.07(s,1h),7.63(d,j=7.7hz,1h),7.58(s,1h),7.49(d,j=8.5hz,1h),7.29(dd,j=1.9hz,8.9hz,1h),6.79(d,j=8.8hz,1h),4.84(s,2h),3.37(m,1h),2.96(s,3h),2.80(s,3h),2.32(t,j=7.6hz,2h),2.06(s,3h),2.04(s,3h),1.98(m,1h),1.70(m,1h),1.34(s,9h).
[0220]
13
c nmr(600mhz,dmso-d6,δppm):172.94,172.60,170.09,168.93,168.10,158.93,157.46,156.93,143.57,136.99,134.49,131.48,129.18,128.30,127.60,117.09,115.79,115.53,113.87,112.66,93.21,80.07,68.26,55.12,36.11,35.52,30.09,28.29,24.46,23.62,23.40.
[0221]
esi-ms:m/z 756.2460(m+h)
+
[0222]
向化合物101(100mg,0.13mmol,1equiv)的dmf(3ml)溶液中,加入化合物93(37.5mg,0.13mmol,1equiv)和dipea(50.3mg,0.39mmol,3equiv),升温至60℃反应1h。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯甲烷/甲醇=20/1至5/1),得到化合物102(97.0mg,白色固体,产率85.0%);
[0223]1h nmr(600mhz,cd3od,δppm):8.75(s,2h),8.42(t,j=9.9hz,2h),8.01(s,2h),7.65(d,j=8.9hz,1h),7.58(d,j=2.0hz,1h),7.42(dd,j=2.1hz,8.2hz,1h),7.30(dd,j=2.1hz,9.0hz,1h),7.15(dd,j=1.9hz,8.3hz,1h),6.88(d,j=8.9hz,1h),4.79(m,3h),2.97(s,3h),2.84(s,3h),2.46(t,j=7.6hz,2h),2.34(m,1h),2.12(s,3h),2.10(s,3h),2.01(s,1h),1.40(s,9h).
[0224]
13
c nmr(600mhz,cd3od,δppm):172.73,171.22,170.46,170.11,169.26,165.92,164.33,158.69,157.26,156.59,147.72,147.61,144.69,141.50,136.25,134.53,131.21,129.51,128.28,127.28,117.83,117.09,116.14,115.04,114.33,109.26,109.01,92.76,80.58,68.57,54.46,34.84,34.51,29.40,27.01,22.62,22.39,21.68.
[0225]
esi-ms:m/z 879.2580(m+h)
+
[0226]
向化合物102(100mg,0.11mmol,1equiv)的dmf(3ml)溶液中,加入tfa(12.9mg,0.11mmol,1equiv),升温至60℃反应1h。旋蒸浓缩反应液,中压制备flash硅胶柱分离(二氯
甲烷/甲醇=20/1至5/1),得到化合物103(68.8mg,白色固体,产率76.1%);
[0227]1h nmr(600mhz,cd3od,δppm):8.72(s,1h),8.42(t,j=6.7hz,1h),7.97(s,1h),7.92(s,1h),7.61(d,j=8.7hz,1h),7.56(s,1h),7.38(d,j=8.6hz,1h),7.25(d,j=7.5hz,1h),7.10(d,j=8.6hz,1h),6.84(d,j=8.6hz,1h),4.73(s,2h),4.62(d,j=4.5hz,1h),2.93(s,3h),2.80(s,3h),2.38-2.35(m,2h),2.20-2.07(m,8h).
[0228]
esi-hrms m/z calculated for c
35
h
37
brfn
10
o
8+
823.1958,found 823.1953[m+h]
+
.
[0229]
制备例7.化合物[
18
f]75、[
18
f]83和[
18
f]84的f-18标记和分离纯化[
18
f]fetos的合成:
[0230][0231]
将15mg kryptofix 222溶于0.7ml无水乙腈中,1mg k2co3溶于0.3ml水中,混合后用其将俘获在qma柱上的
18
f-淋洗至反应瓶中,100℃条件下用n2流将反应瓶中的溶剂吹干,然后向其中加入0.5ml无水乙腈,再次吹干,该过程反复进行三次,保证充分除去反应瓶中的水分;将第一步标记前体1,2-双甲苯氧基乙烷(4mg)的无水乙腈溶液(0.3ml)迅速加入到上述反应瓶中,密封,110℃条件下反应10min。反应结束后,以乙腈:水=1:1为流动相进行radio-hplc分离纯化,流速为4ml/min,波长为254nm,c18反相半制备柱(agela technologies,5μm,10
×
250mm)。产物2-[
18
f]氟乙基甲苯磺酸酯([
18
f]fetos)的保留时间为12分钟。
[0232]
[
18
f]75的合成:
[0233][0234]
将上一步的产物2-[
18
f]氟乙基甲苯磺酸酯([
18
f]fetos)的hplc接收液收集到100ml无菌小瓶中,加入50ml水。将稀释的产物溶液挂载到sep-pak c-18柱上,n2气流吹干sep-pak c-18小柱,用3ml乙醚洗脱sep-pak c-18柱上的产物到无菌小瓶中,40℃下用氮气流吹干。将第二步标记前体(化合物74,4mg)的乙腈(0.4ml)溶液加入其中,再加入无水碳酸钾(1mg)后混合均匀,120℃下反应20分钟。通过半制备柱(agela technologies,5μm,10
×
250mm)液相分离产物。如图1所示,化合物[
18
f]75液相保留时间为22.8分钟,并同化合物75液相共注射分析确认了放射性化合物[
18
f]75的准确性。
[0235]
[
18
f]83、[
18
f]84的合成:
[0236][0237]
[
18
f]83的合成:
[0238]
将15mg kryptofix 222溶于0.7ml无水乙腈中,1mg k2co3溶于0.3ml水中,然后将两者均匀混合配制成1.0ml kryptofix 222/k2co3淋洗液;用该淋洗液将俘获在qma柱上的
18
f-淋洗至反应瓶中,100℃条件下用n2流将反应瓶中的溶剂吹干,然后向其中加入0.5ml无水乙腈,再次吹干,该过程反复进行三次,保证充分除去反应瓶中的水分;将第一步标记前体82(4mg)的无水dmf溶液(0.3ml)迅速加入到上述反应瓶中,密封,110℃条件下反应20min。反应结束后,c18反相半制备柱(agela technologies,5μm,10
×
250mm)分离产物,得到放射性标记的化合物[
18
f]83。
[0239]
[
18
f]84的合成:
[0240]
向radio-hplc液相接出的化合物[
18
f]83产物,加入1m的盐酸(200μl),升温至100℃反应20min。c18反相半制备柱(agela technologies,5μm,10
×
250mm)分离产物,得到放射性标记的化合物[
18
f]84。
[0241]
如图2所示,化合物[
18
f]83的液相保留时间为8.4分钟,并同化合物83液相共注射分析确认了放射性配体[
18
f]83的准确性。
[0242]
如图3所示,化合物[
18
f]84的液相保留时间为3.4分钟,并同化合物84液相共注射分析确认了放射性配体[
18
f]84的准确性。
[0243]
制备例8.化合物[
18
f]102和[
18
f]103的f-18标记和分离纯化
[0244]
合成路线如下:
[0245]
[0246][0247]
室温下向6-氯烟酸(化合物85)(15.0g,95.5mmol,1equiv)的1,4-二氧六环(500ml)溶液中,加入2,3,5,6-四氟苯酚(化合物86)(15.8g,95.5mmol,1equiv)和二环己基碳二亚胺dcc(21.6g,105.0mmol,1.1equiv),室温下搅拌过夜。反应结束后将副产物dcu过滤出来,滤液旋蒸浓缩,中压制备flash硅胶柱分离(石油醚/乙酸乙酯=20/1至5/1),得到化合物87(22.7g,白色固体,产率78.2%).
[0248]1h nmr(400mhz,cdcl3,δppm):9.18(s,1h),8.41(d,j=8.2hz,1h),7.54(d,j=8.3hz,1h),7.12(m,1h).
[0249]
室温下向化合物87(0.5g,1.5mmol,1equiv)的无水thf(3ml)溶液中,加入2m三甲胺(化合物88)的四氢呋喃溶液(2.5ml,5.0mmol,3.3equiv),室温下搅拌过夜。过滤收集沉淀物,并用二氯甲烷(50ml*2)洗涤。沉淀物为化合物89,不需要进一步提纯,不影响之后反应。将沉淀物悬浮在二氯甲烷(5ml)溶液中,超声5分钟。向其加入三氟甲磺酸三甲基硅酯(化合物90)(0.5ml,2.6mmol),室温下剧烈搅拌过夜。过滤,收集浓缩滤液,析出固体,用乙醚(50ml*3)洗涤固体,得到化合物91n
+
(0.2g,白色固体,产率28.5%).
[0250]1h nmr(400mhz,cd3od,δppm):9.40(s,1h),8.93(d,j=8.5hz,1h),8.28(d,j=8.6hz,1h),7.58(m,1h),3.74(s,9h).
[0251]
19
f nmr(400mhz,cd3od,δppm):-80.06(s,3f),-140.90(m,4f).
[0252]
首先将2mg kryptofix 222溶于0.7ml无水乙腈中,2mg khco3溶于0.3ml水中,然后将两者均匀混合配制成1.0ml kryptofix 222/k2co3淋洗液;用该淋洗液将俘获在qma柱上的
18
f-淋洗至反应瓶中,100℃条件下用n2气流将反应瓶中的溶剂吹干,然后向其中加入
0.5ml无水乙腈,再次吹干,该过程反复进行三次,保证充分除去反应瓶中的水分;将标记前体化合物91n
+
(8mg)的无水乙腈/无水叔丁醇(2/8 0.3ml)迅速加入到上述反应瓶中,密封,40℃条件下反应10min。反应结束后,加乙腈(1ml)稀释,抽取到注射器中通过mcx plus sep-pak打入无菌青霉素小瓶中,60℃下n2气流吹干,即纯的放射性标记的化合物[
18
f]93。
[0253]
将2mg的前体(按照制备例6方法制备的化合物101)溶解于无水dmso(0.5ml)中,加入到制备好的化合物[
18
f]93青霉素小瓶中,再加入dipea(20μl),密封,60℃下加热20min,反应完后加水(10ml)猝灭,稀释液打入活化好c-18小柱,空气吹干,再用甲醇(1ml)通过该c-18小柱将产物洗出,可直接进radio-hplc纯化,得到放射性标记的化合物[
18
f]102。
[0254]
将从radio-hplc接出的化合物[
18
f]102纯品转移到无菌青霉素小瓶中,加入三氟乙酸(300μl),密封,60℃下加热20min,反应完后加水(10ml)猝灭,稀释液打入活化好c-18小柱,空气吹干,再用甲醇(1ml)通过该c-18小柱将产物洗出,可直接进radio-hplc纯化,得到放射性标记的化合物[
18
f]103。
[0255]
如图4所示,化合物[
18
f]102的液相保留时间为5.5分钟,并同化合物102液相共注射分析确认了放射性配体[
18
f]102的准确性。
[0256]
如图5所示,化合物[
18
f]103的液相保留时间为3.7分钟,并同化合物103液相共注射分析确认了放射性配体[
18
f]103的准确性。
[0257]
实验例1.体外fak酶活性抑制实验和不同激酶活性选择性抑制实验
[0258]
以前述制备例1-6制备的10种化合物作为本实验的待评估标准品。
[0259]
本测试使用cisbio公司的均相时间分辨的荧光共轭能量转移(方法)进行活性检测。fak激酶购自carna公司;检测试剂盒购自cisbio公司;检测板和多功能酶标仪购自perkin elmer公司。在检测板中,将酶、生物素标记的多肽底物、atp以及检测化合物混合,孵育反应。化合物共11个浓度,最终体系浓度从10μm至0.17nm。用10μl的缓冲液反应体系(50mm hepes ph 7.5,1mm edta,10mm mgcl2,0.01%brij-35,25nm seb,1mm dtt,0.7nm fak,1μm biotin-tk peptide,25μm atp)在23℃下孵育90分钟。加入10μl的终止溶液(20mm edta,0.67nm tk抗体,50nm xl-665),在23℃孵育60分钟,envision读数。将仪器读取的数据计算出化合物的抑制率,然后运用idbs的xlfit5中mode 205计算出ic
50
值。
[0260]
表1各化合物的体外fak酶活性抑制实验结果
[0261]
[0262][0263][0264]
gsk-2256098(上海药明康德新药开发有限公司)是一个在二期临床实验的fak抑制剂,以它为阳性对照药进行fak激酶体外抑制活性实验,结果如表1所示。表1的结果显示化合物79、77、75、95和102的fak-ic
50
均比阳性对照药gsk-2256098低,说明这些化合物对fak的抑制作用优于gsk-2256098;
[0265]
此外,在药物研发领域,根据里宾斯基规则,通常要求化合物的脂水分配系数的对数值(logp)在-2到5之间,其中logp在0-2左右生物体内代谢过程中会有更高的生物利用
度,本申请通过本领域常用的acd labs pro v10软件的clogp模块,导入化合物结构直接计算得到clogp,与logp相近。从结果中可以看出,本申请的化合物的clogp均在-2到5之间,大部分在0-2之间,且较阳性对照药gsk-2256098低,说明本申请的化合物有更好的口服利用度。
[0266]
实验例2.f-18放射性药物在荷s180肿瘤小鼠体内的生物分布实验
[0267]
2.1动物模型建立
[0268]
无菌条件下,酒精消毒正常雌性昆明种小鼠(18-22g)腋下皮肤;取生长良好的s180腹水细胞(购自上海择叶生物科技有限公司,货号zy-m019),以生理盐水稀释至细胞浓度为(1-5)
×
107个/ml,取0.1ml接种于雌性昆明种小鼠腋下皮下。
[0269]
等到肿瘤尺寸长到直径为0.5-0.8cm(大约需要一周时间)的大小可用于生物分布实验,即为s180荷瘤小鼠。
[0270]
2.2动物放射性体内分布实验
[0271]
将经过hplc纯化的制备例7制备的[
18
f]75、[
18
f]83和[
18
f]84(10μci,溶于0.1ml生理盐水,含5%dmso)通过尾静脉注射的方式注入s180荷瘤小鼠体内(18-22g,雌性,n=5),分别在5min,15min,30min,60min,120min时将小鼠断头处死,解剖取出血、脑、心、肝、脾、肺、肾、肌肉、骨、肠、胃、s180瘤和尾,称量各个器官的湿重并用γ-counter测定其计数,每个组织的摄取情况最终以%id/g表示,%id/g=id/g
÷
1%,其中id/g=组织的放射性计数(counts)
÷
组织质量(mg),1%=每个时相1%id的平均值-尾部放射性计数/100。
[0272]
2.3结果讨论
[0273]
[
18
f]75在s180荷瘤小鼠体内的生物分布数据见表2;[
18
f]83在s180荷瘤小鼠体内的生物分布数据见表3;[
18
f]84在s180荷瘤小鼠体内的生物分布数据见表4;[
18
f]102在s180荷瘤小鼠体内的生物分布数据见表5;[
18
f]103在s180荷瘤小鼠体内的生物分布数据见表6。
[0274]
表2化合物[
18
f]75在s180荷瘤小鼠(18-22g)体内的分布情况
a
(logp 0.2,ic
50 3.7nm)
[0275]
[0276]
a
表中数据为三次测量的平均值
±
标准偏差;
[0277]
表3化合物[
18
f]83在s180荷瘤小鼠(18-22g)体内的分布情况
a
(logp 0.7,ic
50 108nm)
[0278][0279]
a
表中数据为三次测量的平均值
±
标准偏差;
[0280]
表4化合物[
18
f]84在s180荷瘤小鼠(18-22g)体内的分布情况
a
(logp 0.3,ic
50 36.2nm)
[0281]
[0282][0283]
a
表中数据为三次测量的平均值
±
标准偏差;
[0284]
从表2-4中可以看出,本申请的f-18标记的各种化合物在静脉注射后,15分钟内(化合物[
18
f]75)或30分钟内(化合物[
18
f]83,化合物[
18
f]84),在肿瘤中发生了明显富集,其在瘤中的含量逐渐增多。
[0285]
静脉注射后,由于核素的衰变,各化合物在各脏器内的分布整体为下降趋势,但是药物在瘤中和在其他脏器中的含量比均呈上升趋势,说明相比于其他脏器,药物在肿瘤中的含量是逐渐增加的,也说明药物在肿瘤中发生了相对富集。
[0286]
表2-表4的结果表明本发明的上述放射性标记物在s180荷瘤小鼠体内具有理想的生物分布,同样也为这些化合物今后能开发成放射性药物进行肿瘤的早期诊断研究,提供了较为充分的实验依据。
[0287]
实验例3.f-18放射性药物在荷a549荷瘤裸鼠体内分布现象实验
[0288]
向a549(人非小细胞肺癌细胞)荷瘤裸鼠(购买自北京维通利华,品系代码403,专业名称crl:nu-foxn1nu)尾静脉注射实施例7制备的化合物[
18
f]75(10μci,溶于0.1ml生理盐水,含5%dmso),30分钟时,小鼠用含有1.5%异氟烷的空气(气流速度约1.5ml/min)进行麻醉,并采用superargus型小动物pet/ct(sociedadde electromedicina y calidad,s.a.)进行静态扫描(时相:30min,采集10min),获取的数据由sedecal reconstruction软件进行图像重建,图像由mmwks superargus软件进行导出,小鼠体内化合物[
18
f]75的分布结果如图6所示。
[0289]
从图6可以看出,[
18
f]75在肿瘤区有明显的摄取(右侧背部,图6中箭头所示),可以作为肿瘤显像剂。此外,发明人发现[
18
f]75在肠区也有明显的摄取(图6中虚线矩形内),不限于任何理论,发明人认为,化合物[
18
f]75可能主要通过肠代谢,因此在肠中有较高的富集。
[0290]
实验例4[
18
f]75摄取阻断实验
[0291]
为了验证本申请的化合物对fak的靶向作用,采用vs-6063(cas:1073160-26-5,上海舒恩医药)进行[
18
f]75的摄取阻断实验。
[0292]
取s180荷瘤小鼠分成两组,分别为抑制组和未抑制组,尾静脉注射实施例7制备的化合物[
18
f]75(10μci,溶于0.1ml生理盐水,含5%dmso),其中抑制组小鼠提前一小时尾静脉注射vs-6063(33mg/kg);注射化合物[
18
f]75后,分别在第15分钟、30分钟处死小鼠,并采用实验例2的方法检测化合物[
18
f]75在肿瘤中的摄取情况,以%id/g表示,结果如图7所示。
[0293]
从图7中可以看出,抑制组在第15分钟和第30分钟,肿瘤对化合物[
18
f]75的摄取相比于未抑制组均显著降低(*p<0.05,n=6)。vs-6063具有较高的fak靶向性抑制作用,尾静
脉注射vs-6063后化合物[
18
f]75的摄取受抑制,说明化合物[
18
f]75也具有fak靶向性。
[0294]
综上,实验例1说明本申请的化合物对fak具有一定的抑制作用,因此可以用于制备肿瘤治疗药物;实验例2和实验例3说明本申请的化合物能够在肿瘤中富集,实验例4说明本申请是化合物具有fak靶向性,因此本申请的化合物通过靶向fak富集于肿瘤中,一方面进一步说明本申请的化合物能够用于制备肿瘤治疗药物,另一方面还说明本申请的化合物还能够用于制备肿瘤诊断显像剂。
[0295]
以上所述仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内所作的任何修改、等同替换、改进等,均包含在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1