一种从植物组织样本中同时提取DNA、RNA和蛋白的试剂盒以及通用方法与流程
一种从植物组织样本中同时提取dna、rna和蛋白的试剂盒以及通用方法
技术领域
1.本发明属于生物技术领域,具体涉及一种从植物组织样本中同时提取dna、rna和蛋白的试剂盒以及通用方法。
背景技术:
2.为了全面了解药用植物样本分化、发育和调控的情况,通常需要对生物样本进行基因组、转录组以及蛋白组研究的综合分析,因此从药用植物组织样本中有效提取dna、rna和蛋白质等生物分子至关重要。而药用植物样本通常是样本量较少的珍稀样本,如果能从同一个组织样本中同时提取dna、rna和蛋白质等生物分子,则能够大量地节省时间并减少成本,对生物样本也能更有效地利用。其次,同时对同一来源的生物样本进行基因组、转录组以及蛋白组的分析,其相关性研究也不易受到组织异质性的干扰。
3.但是现有的技术方法通常着眼于采用单独的组织样本对dna、rna和蛋白等生物分子进行分别提取。此外,对药用植物进行基因组、转录组以及蛋白组研究对所提取的生物分子的质量要求较高,对所提取的生物分子的纯度和完整性都有较高的要求,故从药用植物组织中同时提取高质量的dna、rna和蛋白具备一定的困难性。
4.目前针对植物dna、rna和蛋白质同时提取的方法研究较少。qiagen、omega以及clontech等公司有开发一些商业试剂盒来用于dna、rna和蛋白质的同时提取。此外,一些以有机试剂抽提为基础的方法也应用于dna、rna和蛋白质的共提取。
5.qiagen和tiangen(天根生化科技有限公司)已经开发出从同一组织样本中同时提取dna、rna和蛋白的试剂盒,然而其试剂盒均是适用于从人源或动物细胞或者组织中同时提取dna、总rna和总蛋白,并不适用于植物组织样本的提取。
6.clontech针对从同一组织中同时提取dna、rna和蛋白液开发出了相应的试剂盒。该试剂盒除人源或动物细胞或者组织外,也能以植物组织样本为起始材料。由于其所采用的方法是对核酸吸附柱结合的dna和rna混合液进行逐步洗涤然后洗脱从而达到分离dna和rna的目的,并采用rdna酶对残余的dna进行消化以保证rna的纯度。此方法造成提取的dna和rna产量偏低,且逐步洗涤如果不完全也会造成提取的dna和rna纯度偏低,从而需要在进行后续分析实验之前进行进一步的酶解消化。
7.采用商业试剂盒同时提取植物组织样本中的dna、rna和蛋白质,其成本通常相对较高。而且商业试剂盒通常是通用型试剂盒,多适用于原核生物细胞、动物细胞、动物组织等样本类型的提取。而药用植物组织样本通常成分比较复杂,其通常含有如多酚类、多糖类、萜类化合物等多种次级代谢产物,这些次级代谢产物给生物分子的提取和纯化带来极大的干扰。因此需要对试剂盒进行多次方法摸索和优化才能用于从药用植物组织样本中同时提取dna、rna和蛋白质,而且提取效果往往不佳,提取的核酸和蛋白分子可能含量较低或者纯度不高,因此会对下游的pcr扩增、转录组分析以及蛋白检测造成影响。
8.其次以有机试剂抽提为基础的提取法通常涉及如苯酚、氯化锂等大量毒性或者刺
激性试剂的使用,其操作过程比较繁琐、操作耗时较长、提取效率较低。而核酸提取往往以沉淀法为基础,涉及异丙醇沉淀和乙醇清洗等步骤,这些过程操作步骤较多,而且对操作人员的要求较高,因为不当操作可能会使提取的核酸遭受有机试剂的污染或者由于回收不完全造成提取产物的含量较低。
技术实现要素:
9.本发明的目的是提供一种从植物组织样本中同时提取dna、rna和蛋白的试剂盒以及通用方法。
10.本发明提供了一种试剂盒,包括提取液、rna沉淀溶解液和蛋白裂解液;
11.提取液含有如下组分:表面活性剂和变性剂;
12.rna沉淀溶解液含有如下组分:表面活性剂和金属离子螯合剂;
13.蛋白裂解液含有如下组分:表面活性剂、金属离子螯合剂和可溶性盐。
14.所述提取液中的所述表面活性剂可为阳离子型表面活性剂、阴离子型表面活性剂、两性离子型表面活性剂或非离子型表面活性剂。具体的,所述表面活性剂为ctab、triton x-100、tween 20、tween 40、tween 60、chaps和np-40中任意一种或者任意几种的组合。ctab,全称为十六烷基三甲基溴化铵。chaps,全称为3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate。np-40,全称为nonyl phenoxypolyethoxylethanol。在高离子强度的溶液里,表面活性剂与蛋白质、多酚和多糖类成分形成复合物沉淀,经过氯仿抽提,核酸溶解于水相之中,从而分离出核酸。
[0015]
所述提取液中的所述变性剂可为高离液盐。所述变性剂具体可为盐酸胍或异硫氰酸胍。变性剂可以使细胞膜裂解和蛋白质变性。
[0016]
所述提取液中还可包括促进核酸分子提取的成分。促进核酸分子提取的成分可为吸附剂和/或还原剂。吸附剂可以结合并去除多酚类物质。还原剂能够保持环境中的还原性从而防止植物组织中的酚类物质被氧化(酚类物质被氧化后,与核酸大分子发生不可逆结合从而影响核酸分子提取)。所述吸附剂为高分子两性聚合物。具体的,所述吸附剂可为pvp或pvpp。所述还原剂为巯醇类化合物。具体的,所述还原剂可为dtt、β-巯基乙醇或tcep。dtt,全称为二硫苏糖醇(dithiothreitol)。β-巯基乙醇,全称为2-mercaptoethanol(βme)。tcep,全称为tris(2-carboxyethyl)phosphine,三(2-羧乙基)膦。
[0017]
所述提取液还可包括缓冲盐。具体的,所述缓冲盐为tris-hcl、tricine、bicine或hepes。
[0018]
所述提取液中,余量为水。
[0019]
所述提取液中,余量为溶剂。
[0020]
所述提取液采用缓冲液作为溶剂。所述缓冲液为弱碱性缓冲液。具体的,所述缓冲液为tris-hcl缓冲液、tricine缓冲液、bicine缓冲液或hepes缓冲液。缓冲液的ph范围为7.5-9.0。
[0021]
所述提取液含有如下组分:表面活性剂、变性剂、吸附剂、还原剂和缓冲盐。
[0022]
所述提取液中:表面活性剂的浓度为1-6g/100ml,变性剂的浓度为2-8mol/l,吸附剂的浓度为0.5-4g/100ml,还原剂的浓度为10-100mmol/l,缓冲盐的浓度为50-500mmol/l。
[0023]
所述提取液中:表面活性剂的浓度为1%(w/v)-6%(w/v)、变性剂的浓度为2mol/
l-8mol/l、吸附剂的浓度为0.5%(w/v)-4%(w/v)、还原剂的浓度为10mmol/l-100mmol/l、缓冲盐的浓度为50mmol/l-500mmol/l。
[0024]
所述提取液中:所述表面活性剂为ctab和tritonx-100,所述变性剂为盐酸胍或异硫氰酸胍,所述吸附剂为pvp k-30,所述还原剂为β-巯基乙醇或dtt,所述缓冲盐为tris-hcl。
[0025]
所述提取液中:ctab的浓度为1-3g/100ml,tritonx-100的浓度为1-3g/100ml,变性剂的浓度为6-8mol/l,吸附剂的浓度为1-3g/100ml,还原剂的浓度为13-16mmol/l,缓冲盐的浓度为90-110mmol/l。
[0026]
所述提取液是采用如下方法制备得到的:1g ctab粉末、1g pvp k-30粉末、0.35mol盐酸胍、1g tritonx-100、2.5ml tris-hcl(ph7.5、2m),加depc处理的水至50ml,使用前加入β-巯基乙醇并使其浓度为14.3mmol/l。
[0027]
所述提取液中:ctab的浓度为1-3g/100ml,tritonx-100的浓度为1-3g/100ml,变性剂的浓度为6-8mol/l,吸附剂的浓度为1-3g/100ml,还原剂的浓度为8-12mmol/l,缓冲盐的浓度为90-110mmol/l。
[0028]
所述提取液是采用如下方法制备得到的:1g ctab粉末、1g pvp k-30粉末、0.35mol异硫氰酸胍、1g tritonx-100、2.5ml tris-hcl(ph7.5、2m),加depc处理的水至50ml,使用前加入dtt并使其浓度为10mmol/l。
[0029]
所述rna沉淀溶解液中的所述表面活性剂可为阴离子型表面活性剂。具体的,所述表面活性剂可为sds或sarcosyl。sds,全称为十二烷基硫酸钠。sarcosyl,全称为n-月桂肌胺酸钠盐(sodium lauroylsarcosinate)。
[0030]
所述rna沉淀溶解液中的所述金属离子螯合剂可为edta或egta。edta,全称为乙二胺四乙酸,即ethylene diamine tetraacetic acid。egta,全称为乙二醇双(2-氨基乙基醚)四乙酸,即ethylenebis(oxyethylenenitrilo)tetraacetic acid。
[0031]
所述rna沉淀溶解液中还含有高盐成分。所述高盐成分为一价离子盐,可为氯化钠、醋酸钠、氯化钾或醋酸钾。
[0032]
所述rna沉淀溶解液还可包括缓冲盐。具体的,所述缓冲盐为tris-hcl、tricine、bicine、hepes或ches。
[0033]
所述rna沉淀溶解液中,余量为水。
[0034]
所述rna沉淀溶解液中,余量为溶剂。
[0035]
所述rna沉淀溶解液采用缓冲液作为溶剂。所述缓冲液可为碱性缓冲液。具体的,所述缓冲液为tris-hcl缓冲液、tricine缓冲液、bicine缓冲液、hepes缓冲液或ches缓冲液。缓冲液的ph范围为7.5-8.5。
[0036]
所述rna沉淀溶解液含有如下组分:表面活性剂、金属离子螯合剂、一价离子盐和缓冲盐。
[0037]
所述rna沉淀溶解液中:表面活性剂的浓度为0.2-2g/100ml,金属离子螯合剂的浓度为0.5-5mmol/l,一价离子盐的浓度为0.5-5mol/l,缓冲盐的浓度为8-100mmol/l。
[0038]
所述rna沉淀溶解液中:表面活性剂的浓度为0.2%(w/v)-2%(w/v),金属离子螯合剂的浓度为0.5mmol/l-5mmol/l,一价离子盐的浓度为0.5mol/l-5mol/l,缓冲盐的浓度为8mmol/l-100mmol/l。
[0039]
所述rna沉淀溶解液中:所述表面活性剂为sds或sarcosyl,所述金属离子螯合剂为edta,所述一价离子盐为nacl,所述缓冲液为tris-hcl。
[0040]
所述rna沉淀溶解液中:表面活性剂的浓度为0.4-0.6g/100ml,金属离子螯合剂的浓度为0.8-1.2mmol/l,一价离子盐的浓度为0.8-1.2mol/l,缓冲盐的浓度为8-12mmol/l。
[0041]
所述rna沉淀溶解液是采用如下方法制备得到的:12.5ml 4m nacl水溶液、5ml 5%(w/v)sds水溶液、500μl tris-hcl(ph8.0、1m)、100μl edta(ph8.0、0.5m),加depc处理的水至50ml。
[0042]
所述rna沉淀溶解液中:表面活性剂的浓度为0.8-1.2g/100ml,金属离子螯合剂的浓度为0.8-1.2mmol/l,一价离子盐的浓度为0.8-1.2mol/l,缓冲盐的浓度为8-12mmol/l。
[0043]
所述rna沉淀溶解液是采用如下方法制备得到的:12.5ml 4m nacl水溶液、5ml 10%(w/v)sarkosyl水溶液、500μl tris-hcl(ph8.0、1m)、100μl edta(ph8.0、0.5m),加depc处理的水至50ml。
[0044]
所述蛋白裂解液中的所述表面活性剂可为阳离子型表面活性剂、阴离子型表面活性剂、两性离子型表面活性剂或者非离子型表面活性剂。具体的,所述表面活性剂为sds、sarcosyl、triton x-100、tween 20、tween 40、tween 60、chaps和np-40中任意一种或者任意几种的组合。
[0045]
所述蛋白裂解液中的所述金属离子螯合剂可为edta或egta。
[0046]
所述蛋白裂解液中的所述可溶性盐为一价离子盐。具体的,所述可溶性盐为氯化钠、醋酸钠、氯化钾或醋酸钾。
[0047]
所述蛋白裂解液中还可包括抑制蛋白质水解的成分。抑制蛋白质水解的成分可为还原剂和/或蛋白酶抑制剂。所述还原剂为巯醇类化合物。具体的,所述还原剂可为dtt、β-巯基乙醇或tcep。所述蛋白酶抑制剂可为抑肽酶、苯丁抑制素、亮抑酶肽、aebsf、e64和抑胃酶肽a中任意一种或者任意几种的组合(示例性的可为具有所述6种成分的组合商业蛋白酶抑制剂)。
[0048]
所述蛋白裂解液还可包括缓冲盐。具体的,所述缓冲盐为tris-hcl、tricine、bicine、hepes或ches。
[0049]
所述蛋白裂解液中,余量为水。
[0050]
所述蛋白裂解液中,余量为溶剂。
[0051]
所述蛋白裂解液采用缓冲液作为溶剂。所述缓冲液可为碱性缓冲液。具体的,所述缓冲液为tris-hcl缓冲液、tricine缓冲液、bicine缓冲液、hepes缓冲液或ches缓冲液。缓冲液的ph范围为7.5-9.0。
[0052]
所述蛋白裂解液含有如下组分:表面活性剂、金属离子螯合剂、可溶性盐、蛋白酶抑制剂、还原剂和缓冲盐。
[0053]
所述蛋白裂解液中:表面活性剂的浓度为0.5-2g/100ml,金属离子螯合剂的浓度为0.5-5mmol/l,可溶性盐的浓度为50-300mmol/l,蛋白酶抑制剂的体积百分含量为0.1%-2%,还原剂的浓度为0.8-10mmol/l,缓冲盐的浓度为10-100mmol/l。
[0054]
所述蛋白裂解液中:表面活性剂的浓度为0.5%(w/v)-2%(w/v),金属离子螯合剂的浓度为0.5mmol/l-5mmol/l,可溶性盐的浓度为50mmol/l-300mmol/l,蛋白酶抑制剂的浓度为0.1%(v/v)-2%(v/v),还原剂的浓度为0.8mmol/l-10mmol/l,缓冲盐的浓度为
10mmol/l-100mmol/l。
[0055]
所述蛋白裂解液中:所述表面活性剂为sds和triton x-100,所述金属离子螯合剂为edta,所述可溶性盐为nacl,所述还原剂为dtt,所述缓冲盐为tris-hcl。
[0056]
所述蛋白裂解液中:sds的浓度为0.08-0.12g/100ml,tritonx-100的浓度为0.8-1.2g/100ml,金属离子螯合剂的浓度为1-3mmol/l,可溶性盐的浓度为140-160mmol/l,蛋白酶抑制剂的体积百分含量为0.5%-1.5%,还原剂的浓度为0.8-1.2mmol/l,缓冲盐的浓度为90-100mmol/l。
[0057]
所述蛋白裂解液是采用如下方法制备得到的:1.875ml 4m nacl水溶液、200μl edta(ph8.0、0.5m)、2.5ml tris-hcl(ph7.5、2m)、1ml 5%(w/v)sds水溶液、0.5g tritonx-100、50μl 1m dtt水溶液,加depc处理的水至50ml,使用前加入0.5ml商业蛋白酶抑制剂。
[0058]
所述试剂盒还包括洗涤液1和洗涤液2;
[0059]
洗涤液1含有如下组分:金属离子螯合剂、可溶性盐和质子溶剂;
[0060]
洗涤液2含有如下组分:可溶性盐和质子溶剂。
[0061]
所述洗涤液1中的所述金属离子螯合剂可为edta或egta。
[0062]
所述洗涤液1中的所述可溶性盐为一价离子盐。具体的,所述可溶性盐可为氯化钠、醋酸钠、氯化钾或醋酸钾。
[0063]
所述洗涤液1中的所述质子溶剂为水溶性醇类。具体的,所述质子溶剂可为乙醇或异丙醇。
[0064]
所述洗涤液1还可包括缓冲盐。具体的,所述缓冲盐为tris-hcl、tricine、bicine、hepes或ches。
[0065]
所述洗涤液1中,余量为水。
[0066]
所述洗涤液1中,余量为溶剂。
[0067]
所述洗涤液1采用缓冲液作为溶剂。所述缓冲液可为碱性缓冲液。具体的,所述缓冲液为tris-hcl缓冲液、tricine缓冲液、bicine缓冲液、hepes缓冲液或ches缓冲液。缓冲液的ph范围为7.5-9.0。
[0068]
所述洗涤液1含有如下组分:金属离子螯合剂、可溶性盐、质子溶剂和缓冲盐。
[0069]
所述洗涤液1中:金属离子螯合剂的浓度为0.05-2mmol/l,可溶性盐的浓度为1mmol/l-20mmol/l,质子溶剂的体积百分含量为75%-95%,缓冲盐的浓度为1-10mmol/l。
[0070]
所述洗涤液1中:金属离子螯合剂的浓度为0.05mmol/l-2mmol/l,可溶性盐的浓度为1mmol/l-20mmol/l,质子溶剂的浓度为75%(v/v)-95%(v/v),缓冲盐的浓度为1mmol/l-10mmol/l。
[0071]
所述洗涤液1中:所述金属离子螯合剂为edta,所述可溶性盐为nacl,所述质子溶剂为乙醇,所述缓冲盐为tris-hcl。
[0072]
所述洗涤液1中:金属离子螯合剂的浓度为0.05-0.15mmol/l,可溶性盐的浓度为1-3mmol/l,质子溶剂的体积百分含量为85%-95%,缓冲盐的浓度为1-3mmol/l。
[0073]
所述洗涤液1是采用如下方法制备得到的:25μl 4m nacl水溶液、10μl edta(ph8.0、0.5m)、45ml无水乙醇、50μl tris-hcl(ph7.5、2m),加depc处理的水至50ml。
[0074]
所述洗涤液2中的所述可溶性盐为一价离子盐。具体的所述可溶性盐可为氯化钠、醋酸钠、氯化钾或醋酸钾。
[0075]
所述洗涤液2中的所述质子溶剂为水溶性醇类。具体的,所述质子溶剂可为乙醇或异丙醇。
[0076]
所述洗涤液2还可包括缓冲盐。具体的,所述缓冲盐为tris-hcl、tricine、bicine、hepes或ches。
[0077]
所述洗涤液2中,余量为水。
[0078]
所述洗涤液2中,余量为溶剂。
[0079]
所述洗涤液2采用缓冲液作为溶剂。所述缓冲液可为碱性缓冲液。具体的,所述缓冲液为tris-hcl缓冲液、tricine缓冲液、bicine缓冲液、hepes缓冲液或ches缓冲液。缓冲液的ph范围为7.5-9.0。
[0080]
所述洗涤液2含有如下组分:可溶性盐、质子溶剂和缓冲盐。
[0081]
所述洗涤液2中:可溶性盐的浓度为1mmol/l-20mmol/l,质子溶剂的体积百分含量为60%-80%,缓冲盐的浓度为1-10mmol/l。
[0082]
所述洗涤液2中:可溶性盐的浓度为1mmol/l-20mmol/l,质子溶剂的浓度为60%(v/v)-80%(v/v),缓冲盐的浓度为1mmol/l-10mmol/l。
[0083]
所述洗涤液2中:所述可溶性盐为nacl,所述质子溶剂为乙醇,所述缓冲盐为tris-hcl。
[0084]
所述洗涤液2中:可溶性盐的浓度为1-3mmol/l,质子溶剂的体积百分含量为60%-80%,缓冲盐的浓度为1-3mmol/l。
[0085]
所述洗涤液2是采用如下方法制备得到的:25μl 4m nacl水溶液、35ml无水乙醇、50μl tris-hcl(ph7.5、2m),加depc处理的水至50ml。
[0086]
洗涤液1能够将固相基质上非特异性结合的蛋白清洗掉,而洗涤液2能够将固相基质上残留的盐酸胍、氯化钠等盐离子成分清洗掉。最后洗脱液对固相基质上吸附的核酸进行洗脱,从而可以得到高纯度的dna。
[0087]
沉淀部分的rna通过沉淀溶解液进行复溶,通过固相基质硅胶柱对核酸的特异性吸附能力,使核酸分子得到有效释放。洗涤液1能够将固相基质上非特异性结合的蛋白清洗掉,而洗涤液2能够将固相基质上残留的盐酸胍、氯化钠等盐离子成分清洗掉。最后洗脱液对固相基质上吸附的核酸进行洗脱,从而可以得到高纯度的rna。
[0088]
固相基质离心后收集的滤液成分中含有蛋白质,蛋白沉淀后通过蛋白提取液进行重悬,可以得到相应的蛋白成分。
[0089]
所述试剂盒的用途为提取植物材料的dna、rna和蛋白质。
[0090]
本发明还保护以上任一所述试剂盒的应用,所述应用为从植物材料中提取dna、rna和蛋白质。
[0091]
本发明还保护一种采用以上任一所述试剂盒从植物材料中提取dna、rna和蛋白质的方法,包括如下步骤:
[0092]
(1)将植物材料转移至离心管1,加入提取液进行提取;
[0093]
(2)取完成步骤(1)的离心管1,加入有机相,然后离心,将上层和中间层转移至离心管2,下层仍然保存于离心管1;所述有机相为氯仿或者氯仿和其他有机溶剂的混合液;
[0094]
(3)取完成步骤(2)的离心管2,加入核酸沉淀剂,孵育,然后离心;
[0095]
(4)完成步骤(3)后,将上清液转移至dna吸附介质,离心,将滤液转移至所述离心
管1;然后,洗涤所述dna吸附介质,然后离心;然后,在所述dna吸附介质中加入不含核酸酶的水,静置后离心,收集滤液,即为dna溶液;
[0096]
(5)完成步骤(3)后,在装有剩余沉淀的离心管2中加入rna沉淀溶解液,然后加入水溶性醇类(具体的所述水溶性醇类为无水乙醇或异丙醇),混匀后转移至rna吸附介质,离心,将滤液转移至所述离心管1;然后,洗涤所述rna吸附介质,然后离心;然后,在所述rna吸附介质中加入不含核酸酶的水,静置后离心,收集滤液,即为rna溶液;
[0097]
(6)取所述离心管1,加入水溶性有机溶剂(具体的所述水溶性有机溶剂为甲醇、乙腈或丙酮)并孵育,然后离心弃上清;然后用水溶性有机溶剂(具体的所述水溶性有机溶剂为甲醇、乙腈或丙酮)清洗沉淀,然后用水溶性醇类(具体的所述水溶性醇类为无水乙醇或异丙醇)清洗沉淀;然后加入蛋白裂解液进行裂解;然后,离心收集上清液,即为蛋白溶液。
[0098]
示例性的,所述步骤(2)中,所述有机相为氯仿。
[0099]
示例性的,所述步骤(3)中,所述核酸沉淀剂为聚乙二醇类物质,具体可为peg8000。
[0100]
核酸分子沉淀剂通常为强脱水剂氯化锂或者聚乙二醇类。由于氯化锂具有遗传毒性且沉淀核酸分子时往往需要过夜处理,而聚乙二醇类物质对dna具有凝聚效应,因此本发明选择聚乙二醇类物质作为核酸分子沉淀剂。聚乙二醇类物质对dna的凝聚效应,使核酸成分中的dna和rna有效分离。优选的核酸分子沉淀剂为peg 8000。
[0101]
示例性的,所述dna吸附介质为dna吸附柱。
[0102]
示例性的,所述rna吸附介质为rna吸附柱。
[0103]
本发明的实施例中采用的是核酸吸附柱来进行核酸的结合、清洗和最终洗脱。采用具有高吸附能力的96孔板如果能达到同样的核酸吸附要求,那么在通量上会比核酸吸附柱高,在一块板上能实现更多样本的提取。
[0104]
洗涤所述dna吸附介质的方法如下:依次采用洗涤液1和洗涤液2进行洗涤;
[0105]
洗涤所述rna吸附介质的方法如下:依次采用洗涤液1、洗涤液2和洗涤液2进行洗涤。
[0106]
所述植物材料可为粉末状植物材料,例如液氮研磨获得的粉末状植物材料。
[0107]
所述植物材料可为液氮冻存的植物材料。
[0108]
所述植物材料也可以是新鲜的植物材料。
[0109]
所述植物材料包括但不限于植物组织,例如叶片组织、根组织等。
[0110]
作为一种示例,所述方法包括如下步骤:
[0111]
1、取植物材料100mg,加入液氮预冷的研钵中,加入液氮,研磨成均匀粉末。
[0112]
2、将步骤1得到的粉末转移至2ml离心管(称为离心管1)中,加入1ml预热至65℃的提取液,涡旋15s以混匀,然后65℃孵育5min。
[0113]
3、取完成步骤2的离心管1,静置冷却至室温,加入0.2ml氯仿,剧烈摇晃使混合均匀,然后4℃、12000g离心10min(此时分成三层,上层为水相,下层为有机相),将上层和中间层转移至新的2ml离心管(称为离心管2)中,下层仍然保存于离心管1中用于后续蛋白提取。
[0114]
4、取完成步骤3的离心管2,加入30%(w/v)peg8000水溶液(peg8000水溶液的加入体积为离心管2中液体的体积的十分之一),冰上孵育30min,然后4℃、12000g离心10min。
[0115]
5、完成步骤4后,将上清液转移至置于2ml收集管的dna吸附柱中,轻轻合上盖子,
室温孵育1min,随后10000g离心1min,将滤液(称为滤液1)转移至所述离心管1中用于后续蛋白提取;然后,在dna吸附柱中加入500μl洗涤液1,10000g离心1min,弃滤液;然后,在dna吸附柱中加入500μl洗涤液2,10000g离心2min,弃滤液;然后,10000g离心1min以使dna吸附柱的膜彻底干燥;然后,将dna吸附柱转移至新的1.5ml收集管中,在dna吸附柱中加入100μl不含核酸酶的水,室温静置2min,然后10000g离心1min,收集滤液,即为dna溶液,-20℃保存。
[0116]
6、完成步骤4后,在装有剩余沉淀的离心管2中加入300μl 65℃预热的rna沉淀溶解液进行溶解,然后加入450μl无水乙醇,混合均匀,然后将混合液转移至置于2ml收集管的rna吸附柱中,10000g离心1min,将滤液(称为滤液2)转移至所述离心管1用于后续蛋白提取;然后,在rna吸附柱中加入500μl洗涤液1,10000g离心1min,弃滤液;然后,在rna吸附柱中加入500μl洗涤液2,10000g离心1min,弃滤液;然后,在rna吸附柱中加入500μl洗涤液2,10000g离心2min,弃滤液;然后,10000g离心1min以使rna吸附柱的膜彻底干燥;然后,将rna吸附柱转移至新的1.5ml收集管中,在rna吸附柱中加入40μl不含核酸酶的水,室温静置2min,然后10000g离心1min,收集滤液,即为rna溶液,-20℃保存。
[0117]
7、取所述离心管1(其中含有步骤3中的下层、步骤5中的滤液1和步骤6中的滤液2),加入丙酮(丙酮的加入体积为离心管1中液体体积的2.5倍),-20℃孵育30min,然后4℃、10000g离心5min,弃上清;然后,对沉淀进行两次清洗(每次清洗的方法均为:加入1ml丙酮清洗沉淀,然后4℃、5000g离心5min,弃上清);然后,用1ml提前预冷的无水乙醇清洗沉淀(4℃、5000g离心5min,弃上清);然后,用200μl蛋白裂解液重悬沉淀,50℃孵育5min以促进沉淀溶解;然后,16000g离心1min,收集上清液,即为蛋白溶液,-20℃保存。
[0118]
所述植物可为药用植物。
[0119]
所述植物可为稀有植物。
[0120]
示例性的,所述植物可为藿香或独一味。
[0121]
本发明的提取液提供还原性环境,如果提取液不提供还原性的环境,植物组织中富含的多酚类物质会被氧化成醌类物质,从而与核酸大分子发生不可逆的结合,在后续的氯仿抽提过程中造成核酸分子的损失。本发明中,ctab与高浓度的高离液盐的结合使用,胍盐通常具有较强的变性能力,能够促使细胞膜得到有效裂解而且可以使缠绕在dna上的蛋白变性,释放dna。ctab进一步与蛋白、多酚、多糖类成分形成沉淀,使核酸分子能释放出来。核酸沉淀剂的选择:通常方法采用氯化锂作为核酸沉淀剂,但是氯化锂沉淀rna效率较低,耗时较长,使用peg 8000能加快沉淀过程,缩短提取的总时间。适当的缓冲盐浓度ph条件,能够提供缓冲环境,防止核酸被降解破坏,从而保证核酸的稳定性。
[0122]
本发明提供了一种适用于从植物组织样本中同时提取dna、rna和蛋白质的通用技术方法。采用实验室的常规试剂配制提取液和洗涤液,成本低廉,且能够高效快捷地同时从植物组织样本中提取高质量高纯度的dna、rna和蛋白质,并减少了有毒试剂的使用以及耗时劳力的操作。同时本发明的方法能够有效去除植物组织样本中常见的多糖类、多酚类等次级代谢产物的影响,所提取的dna、rna和蛋白质质量较高,具有较高的产量且完整性较好,能够满足cdna文库构建、转录组分析、pcr扩增、基因组研究和蛋白分析等下游分子实验分析的要求。由于其提取的dna、rna和蛋白等生物大分子来源于同一组织样本,如果作为一种常规的提取手段进行广泛应用,其与目前流行的从单个样本中分别提取纯化的技术相
比,简化了提取流程,节省了操作时间,提取效率更高效。而且此方法尤其适用于珍稀样本中生物大分子的提取,如有些药用植物样本稀少,因此这类珍稀样本采用同一样本同时提取生物大分子,可以减少样本损耗。其次,同时对同一来源的生物样本进行基因组、转录组以及蛋白分析,也避免了组织异质性导致的结果差异。因此,此方法将在珍稀样本的跨组学相关性研究方面具有广泛的应用前景。
[0123]
本发明对于同时对生物样本进行基因组、转录组以及蛋白组综合分析具有重要的作用,尤其是对于珍稀的药用植物样本,可以节省样本材料的同时对植物样本进行综合分析,同时避免了不同来源组织样本之间可能存在的异质性。
附图说明
[0124]
图1为实施例2的方法与对比例1的方法提取的dna的琼脂糖凝胶电泳结果比较图。m:分子量标准(λ-hindⅲdigest dna marker);泳道a-c为同一试验组的3个重复。
[0125]
图2为实施例2从藿香叶片组织中提取的基因组dna用于扩增pts基因的pcr产物的琼脂糖凝胶电泳图。m:分子量标准(dl2000 dna marker);泳道a-c对应提取的dna的3个重复的pcr产物。
[0126]
图3为实施例2的方法与对比例1的方法提取的rna的琼脂糖凝胶电泳结果比较图。m:分子量标准(dl2000 dna marker);泳道a为对比例1的方法提取的rna;泳道b为实施例2的方法提取的rna。
[0127]
图4为实施例2的方法与对比例1的方法提取的rna经过反转录合成第一链后扩增actin基因的pcr产物的琼脂糖凝胶电泳结果比较图。m:分子量标准(dl2000 dna marker);泳道a为对比例1的方法提取的rna的rt-pcr结果图;泳道b为实施例2的方法提取的rna的rt-pcr结果图。
[0128]
图5为实施例2的方法与对比例1的方法提取的蛋白的sds-page电泳结果比较图。m:分子量标准(premixed protein marker(broad));泳道a为实施例2的方法提取的蛋白的结果图;泳道b为对比例1的方法提取的蛋白的结果图。
[0129]
图6为实施例4提取的dna的琼脂糖凝胶电泳结果图。m1:分子量标准(λ-hindⅲdigest dna marker);m2:分子量标准(dl2000 dna marker)。
[0130]
图7为从实施例4提取的dna中扩增chs基因的pcr产物的琼脂糖凝胶电泳图。m:分子量标准(50bp dna ladder)。
[0131]
图8为实施例4提取的rna的琼脂糖凝胶电泳结果图。m1:分子量标准(dl2000 dna marker);m2:分子量标准(λ-hindⅲdigest dna marker)。
[0132]
图9为实施例4提取的rna经过反转录合成第一链后扩增actin基因的pcr产物的琼脂糖凝胶电泳图。m1:分子量标准(50bp dna ladder);m2:分子量标准(dl2000 dna marker)。
[0133]
图10为实施例4提取的的蛋白的sds-page电泳结果图。m:分子量标准(premixed protein marker(broad))。
[0134]
图11为实施例6的方法提取的dna与实施例4的方法提取的dna的琼脂糖凝胶电泳结果对比图。m1:分子量标准(λ-hindⅲdigest dna marker);m2:分子量标准(dl2000 dna marker)。
[0135]
图12为实施例6的方法提取的rna与实施例4的方法提取的rna的琼脂糖凝胶电泳结果对比图。m1:分子量标准(dl2000 dna marker);m2:分子量标准(λ-hindⅲdigest dna marker)。
具体实施方式
[0136]
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
[0137]
下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。实施例中,%(w/v)指的是100ml溶液所含有的溶质(g),%(v/v)指的是l/l。
[0138]
ctab,即十六烷基三甲基溴化铵,购自bbi。pvp k-30,即聚乙烯吡咯烷酮,购自bbi。盐酸胍,购自bbi。异硫氰酸胍,购自sigma。氯化钠,购自bbi。sarcosyl(sodium lauroylsarcosinate,n-月桂肌胺酸钠盐),购自sigma。tritonx-100,即聚乙二醇辛基苯基醚,购自bbi。tris-hcl(ph7.5、2m),购自sigma。depc,即焦碳酸二乙酯。depc处理的水,购自ambion。β-巯基乙醇(14.3m),购自amresco。sds,即十二烷基硫酸钠,购自bbi。tris-hcl(ph8.0、1m),购自sigma。edta即乙二胺四乙酸。edta(ph8.0、0.5m),购自bbi。dtt即二硫苏糖醇,购自生工生物工程(上海)有限公司。商业蛋白酶抑制剂,即“halt
tm protease inhibitor cocktail,edta-free(100x)”,购自thermofisher scientific。peg8000,购自碧云天。
[0139]
dna吸附柱:dna mini columns(dnacol-02,购自omega bio-tek)。
[0140]
rna吸附柱:rna spin columns(rnacol-02,购自omega bio-tek)。
[0141]
实施例1、试剂的制备
[0142]
提取液的制备:1g ctab粉末、1g pvp k-30粉末、0.35mol盐酸胍、1g tritonx-100、2.5ml tris-hcl(ph7.5、2m),加depc处理的水至50ml,使用前加入β-巯基乙醇并使其浓度为14.3mmol/l。
[0143]
rna沉淀溶解液的制备:12.5ml 4m nacl水溶液、5ml 5%(w/v)sds水溶液、500μl tris-hcl(ph8.0、1m)、100μl edta(ph8.0、0.5m),加depc处理的水至50ml。
[0144]
洗涤液1的制备:25μl 4m nacl水溶液、10μl edta(ph8.0、0.5m)、45ml无水乙醇、50μl tris-hcl(ph7.5、2m),加depc处理的水至50ml。
[0145]
洗涤液2的制备:25μl 4m nacl水溶液、35ml无水乙醇、50μl tris-hcl(ph7.5、2m),加depc处理的水至50ml。
[0146]
蛋白裂解液的制备:1.875ml 4m nacl水溶液、200μl edta(ph8.0、0.5m)、2.5ml tris-hcl(ph7.5、2m)、1ml 5%(w/v)sds水溶液、0.5g tritonx-100、50μl 1m dtt水溶液,加depc处理的水至50ml,使用前加入0.5ml商业蛋白酶抑制剂。
[0147]
实施例2、同时提取藿香组织样本中的dna、rna和蛋白质
[0148]
采用实施例1制备的提取液、rna沉淀溶解液、洗涤液1、洗涤液2和蛋白裂解液。
[0149]
设置三个重复样本,分别命名为样本a1、样本b1和样本c1。每个样本均为藿香(来
源于国家基因库五楼平台栽培地)的新鲜叶片组织。
[0150]
1、取样本100mg,加入液氮预冷的研钵中,加入液氮,研磨成均匀粉末。
[0151]
2、将步骤1得到的粉末转移至2ml离心管(称为离心管1)中,加入1ml预热至65℃的提取液,涡旋15s以混匀,然后65℃孵育5min。
[0152]
3、取完成步骤2的离心管1,静置冷却至室温,加入0.2ml氯仿,剧烈摇晃使混合均匀,然后4℃、12000g离心10min(此时分成三层,上层为水相,下层为有机相),将上层和中间层转移至新的2ml离心管(称为离心管2)中,下层仍然保存于离心管1中用于后续蛋白提取。
[0153]
4、取完成步骤3的离心管2,加入30%(w/v)peg8000水溶液(peg8000水溶液的加入体积为离心管2中液体的体积的十分之一),冰上孵育30min,然后4℃、12000g离心10min。
[0154]
5、完成步骤4后,将上清液转移至置于2ml收集管的dna吸附柱中,轻轻合上盖子,室温孵育1min,随后10000g离心1min,将滤液(称为滤液1)转移至所述离心管1中用于后续蛋白提取;然后,在dna吸附柱中加入500μl洗涤液1,10000g离心1min,弃滤液;然后,在dna吸附柱中加入500μl洗涤液2,10000g离心2min,弃滤液;然后,10000g离心1min以使dna吸附柱的膜彻底干燥;然后,将dna吸附柱转移至新的1.5ml收集管中,在dna吸附柱中加入100μl不含核酸酶的水,室温静置2min,然后10000g离心1min,收集滤液,即为dna溶液,-20℃保存。
[0155]
6、完成步骤4后,在装有剩余沉淀的离心管2中加入300μl 65℃预热的rna沉淀溶解液进行溶解,然后加入450μl无水乙醇,混合均匀,然后将混合液转移至置于2ml收集管的rna吸附柱中,10000g离心1min,将滤液(称为滤液2)转移至所述离心管1用于后续蛋白提取;然后,在rna吸附柱中加入500μl洗涤液1,10000g离心1min,弃滤液;然后,在rna吸附柱中加入500μl洗涤液2,10000g离心1min,弃滤液;然后,在rna吸附柱中加入500μl洗涤液2,10000g离心2min,弃滤液;然后,10000g离心1min以使rna吸附柱的膜彻底干燥;然后,将rna吸附柱转移至新的1.5ml收集管中,在rna吸附柱中加入40μl不含核酸酶的水,室温静置2min,然后10000g离心1min,收集滤液,即为rna溶液,-20℃保存。
[0156]
7、取所述离心管1(其中含有步骤3中的下层、步骤5中的滤液1和步骤6中的滤液2),加入丙酮(丙酮的加入体积为离心管1中液体体积的2.5倍),-20℃孵育30min,然后4℃、10000g离心5min,弃上清;然后,对沉淀进行两次清洗(每次清洗的方法均为:加入1ml丙酮清洗沉淀,然后4℃、5000g离心5min,弃上清);然后,用1ml提前预冷的无水乙醇清洗沉淀(4℃、5000g离心5min,弃上清);然后,用200μl蛋白裂解液重悬沉淀,50℃孵育5min以促进沉淀溶解;然后,16000g离心1min,收集上清液,即为蛋白溶液,-20℃保存。
[0157]
样本a1进行上述步骤,得到的dna溶液命名为dna溶液-a1,得到的rna溶液命名为rna溶液-a1、得到的蛋白溶液命名为蛋白溶液-a1。
[0158]
样本b1进行上述步骤,得到的dna溶液命名为dna溶液-b1,得到的rna溶液命名为rna溶液-b1、得到的蛋白溶液命名为蛋白溶液-b1。
[0159]
样本c1进行上述步骤,得到的dna溶液命名为dna溶液-c1,得到的rna溶液命名为rna溶液-c1、得到的蛋白溶液命名为蛋白溶液-c1。
[0160]
对比例1、
[0161]
一、采用传统ctab法单独提取植物组织样本中的dna
[0162]
设置三个重复样本,分别命名为样本a2、样本b2和样本c2。每个样本均为藿香(来
源于国家基因库五楼平台栽培地)的新鲜叶片组织。
[0163]
1、称取样本100mg于研钵中,加入液氮,快速研磨成粉末,然后转移至2ml离心管中,加入1ml 65℃预热的ctab缓冲液、140μl 20%(w/v)pvp-30水溶液和10μlβ-巯基乙醇,65℃孵育30min。
[0164]
ctab缓冲液:含100mm tris-hcl(ph8.0)、20mm edta(ph8.0)、1.4m nacl、2%(w/v)ctab,余量为水。
[0165]
2、完成步骤1后,取所述离心管,冰上冷却至室温,加入与离心管内液体等体积的氯仿/异戊醇混合液,轻轻摇动10min,室温、12000g离心10min。
[0166]
氯仿/异戊醇混合液:由24体积份氯仿和1体积份异戊醇混合得到。
[0167]
3、完成步骤2后,取上层水相至新的离心管中,加入与离心管内液体等体积的氯仿/异戊醇混合液,轻轻摇动10min,室温、12000g离心10min。
[0168]
4、完成步骤3后,取上层水相至新的离心管中,加入2倍于水相体积的预冷的无水乙醇,吹打混匀,然后-20℃沉淀过夜。
[0169]
5、完成步骤4后,4℃、13600rpm离心20min,取上清液。
[0170]
6、取步骤5得到的上清液,加入500μl预冷的70%(v/v)乙醇水溶液洗涤沉淀1次,吸弃上清,剩余沉淀56℃干燥3min,然后室温干燥2min。
[0171]
7、取步骤6得到的沉淀,加入50μl预热至56℃的灭菌水溶解,加入10μl rnase a(10mg/ml,购自thermofisher scientific),37℃孵育30min以消化rna,得到dna溶液。
[0172]
样本a2进行上述步骤,得到的dna溶液命名为dna溶液-a2。
[0173]
样本b2进行上述步骤,得到的dna溶液命名为dna溶液-b2。
[0174]
样本c2进行上述步骤,得到的dna溶液命名为dna溶液-c2。
[0175]
二、采用传统的sds-酚仿抽提法提取植物组织样本中的rna
[0176]
设置三个重复样本,分别命名为样本a3、样本b3和样本c3。每个样本均为藿香(来源于国家基因库五楼平台栽培地)的新鲜叶片组织。
[0177]
1、称取样本100mg于液氮预冷的研钵内,加入液氮,研磨成粉末,将粉末转移至2ml离心管中。
[0178]
2、完成步骤1后,取所述离心管,加入1ml sds抽提液,涡旋混匀,然后室温静置10min。
[0179]
sds抽提液的制备方法:含0.25m nacl、1%(w/v)sds、50mm tris-hcl(ph7.5),20mm edta(ph8.0),余量为水,用前按照5%(v/v)体积加入β-巯基乙醇。
[0180]
3、完成步骤2后,取所述离心管,4℃、13000g离心15min,收集上清液。
[0181]
4、取步骤3得到的上清液,加入300μl 5m nacl水溶液和500μl氯仿,上下颠倒离心管6-8次以混匀样本,然后4℃、13000g离心5min。
[0182]
5、完成步骤4后,将大约600μl的上层水相转移至新的离心管中,加入与离心管中液体等体积的苯酚/氯仿混合液,上下颠倒离心管6-8次以混匀样本,然后4℃、13000g离心5min。苯酚/氯仿混合液:苯酚和氯仿等体积混合。
[0183]
6、完成步骤5后,将大约400μl的上层水相转移至新的离心管中,加入与离心管中液体等体积的异丙醇,在恒温混匀仪(thermomixer,购自eppendorf)上混匀,室温混匀10min。
[0184]
7、完成步骤6后,将混合液转移到rna吸附柱,然后4℃、13000g离心1min,弃除滤液;然后,向rna吸附柱加入500μl 75%(v/v)乙醇水溶液,然后4℃、13000g离心1min,弃除滤液;然后,向rna吸附柱加入500μl 75%(v/v)乙醇水溶液,然后4℃、13000g离心1min,弃除滤液;然后4℃、13000g离心1min以去除残留乙醇;然后将rna吸附柱装至新的1.5ml离心管,加入50μl不含核酸酶的水溶解,室温静置3min,然后13000g离心1min,收集滤液;再将收集的滤液重新转移至rna吸附柱回溶一次,静置3min,室温、13000g离心1min,收集滤液,即为rna溶液。
[0185]
样本a3进行上述步骤,得到的rna溶液命名为rna溶液-a3。
[0186]
样本b3进行上述步骤,得到的rna溶液命名为rna溶液-b3。
[0187]
样本c3进行上述步骤,得到的rna溶液命名为rna溶液-c3。
[0188]
三、采用植物蛋白提取商业试剂盒提取蛋白
[0189]
设置三个重复样本,分别命名为样本a4、样本b4和样本c4。每个样本均为藿香(来源于国家基因库五楼平台栽培地)的新鲜叶片组织。
[0190]
采用植物蛋白提取商业试剂盒(plant total protein extracion kit,c500053,生工),按说明书操作提取蛋白,得到蛋白溶液。
[0191]
样本a4进行上述步骤,得到的蛋白溶液命名为蛋白溶液-a4。
[0192]
样本b4进行上述步骤,得到的蛋白溶液命名为蛋白溶液-b4。
[0193]
样本c4进行上述步骤,得到的蛋白溶液命名为蛋白溶液-c4。
[0194]
实施例3、提取效果验证
[0195]
一、dna质量验证
[0196]
1、dna的纯度和浓度检测
[0197]
供试溶液:实施例2制备的dna溶液-a1、dna溶液-b1或dna溶液-c1;对比例1的步骤一制备的dna溶液-a2、dna溶液-b2或dna溶液-c2。
[0198]
取1μl供试溶液,采用超微量紫外分光光度计nanodrop 8000(thermofisher scientific)进行dna的纯度和浓度检测。dna的纯度可以由a
260
/a
280
的值进行评估,a
260
/a
280
的值不低于1.8表明dna的纯度较高,a
260
/a
280
的值高于2.0则暗示可能有rna的存在。
[0199]
dna浓度(μg/ml)=od
260
(260nm处吸收值)
×
转换因子(conversion factor);
[0200]
对于双链dna(ds dna),转换因子为:1od
260 unit=50μg/ml。
[0201]
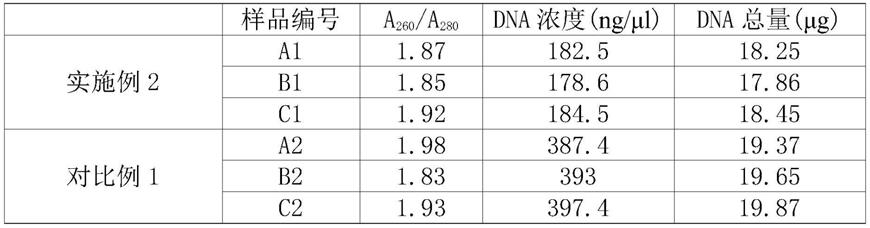
结果见表1。实施例2制备的dna溶液的a
260
/a
280
的值均在1.8-2.0的范围内,表明dna的纯度较高,可以用于后续的实验和分析。与实施例2相比,对比例1制备的dna溶液的dna的总量稍高,但没有明显差异。因此,本发明方法所提取的dna的纯度和产量均能满足下游实验的要求。
[0202]
表1实施例2提取的dna与对比例1提取的dna的质量对比
[0203]
[0204]
2、琼脂糖凝胶电泳检测
[0205]
供试溶液:实施例2制备的dna溶液-a1、dna溶液-b1或dna溶液-c1;对比例1的步骤一制备的dna溶液-a2、dna溶液-b2或dna溶液-c2。
[0206]
取供试溶液(dna含量为150ng),用1.2%含evagreen染料(购自biotium)的琼脂糖凝胶进行电泳检测。电压120v,电泳时间35分钟,电泳后用紫外凝胶成像仪(tanon-1600,上海天能科技有限公司)进行观察和分析。
[0207]
结果见图1。实施例2制备的dna溶液在大于20kb处有一条明显dna条带,且条带完整性良好,未见rna杂质干扰以及dna条带降解。实施例2的方法提取的dna与对比例1的方法提取的dna无差异。
[0208]
3、pcr扩增检测
[0209]
供试溶液:实施例2制备的dna溶液-a1、dna溶液-b1或dna溶液-c1。
[0210]
以供试溶液作为模板,以pts(patchoulol synthase,广藿香醇合成酶)基因(基因序列号:dq355151)横跨外显子6-7的区域为靶序列,进行pcr扩增。
[0211]
正向引物:5
’-
gacaggaatcccattaatat-3’;
[0212]
反向引物:5
’-
ttaatatggaacagggtgaa-3。
[0213]
pcr扩增的反应体系:供试溶液2.5μl、10μm正向引物溶液1μl、10μm反向引物溶液1μl、25μl 2
×
taq pcr mastermix(购自qiagen),加超纯水至50μl。
[0214]
pcr扩增的反应条件:94℃3min;94℃30s、60℃30s、72℃1min,30次循环;72℃5min。
[0215]
pcr扩增产物进行1.2%琼脂糖凝胶电泳,结果见图2。在750bp处明显可见一条清晰的特异性条带,与预期条带的分子量相同,可确定为目的产物片段。说明本方法提取的dna样本完整性良好,可满足下游pcr实验的要求。
[0216]
二、rna质量验证
[0217]
供试溶液:实施例2制备的rna溶液-a1、rna溶液-b1或rna溶液-c1;对比例1的步骤二制备的rna溶液-a3、rna溶液-b3或rna溶液-c3。
[0218]
1、rna的纯度和浓度检测
[0219]
取1μl供试溶液,采用超微量紫外分光光度计nanodrop 8000(thermofisher scientific)进行rna的纯度和浓度检测。rna的纯度可以由a
260
/a
280
进行评估,a
260
/a
280
的值越接近2.0表明rna的纯度较高。a
260
/a
230
的值小于2.0则表明纯化的rna中可能混有盐类、糖类或有机溶剂等成分。
[0220]
rna浓度(μg/ml)=od
260
(260nm处吸收值)
×
转换因子(conversion factor);
[0221]
对于单链rna(ss rna),转换因子为:1od
260 unit=40μg/ml。
[0222]
结果见表2。实施例2制备的rna溶液的a
260
/a
280
的值为2.02,表明rna的纯度较高,可以用于后续的稳定性实验。与实施例2相比,对比例1制备的rna溶液的rna总量虽然稍高,但是纯度偏低。因此,本发明方法所提取的rna的产量虽然略低于传统sds-酚仿抽提法,但差异不显著,而本发明所提取的rna纯度要高于传统sds-酚仿抽提法。
[0223]
2、琼脂糖凝胶电泳检测
[0224]
取供试溶液(rna含量为150ng),用1%含evagreen染料(购自biotium)的琼脂糖凝胶进行电泳检测。电压150v,电泳时间30分钟,电泳后用紫外凝胶成像仪(tanon-1600,上海
天能科技有限公司)进行观察和分析。
[0225]
结果见图3。实施例2制备的rna溶液显示三条明显的条带,分别对应5s、18s和28s,且未见dna的干扰。对比例1制备的rna溶液由于未进行dna消化因此电泳图可见明显的dna条带。因此,本发明提取的rna样本完整性良好,未见dna干扰以及条带降解,可用于后续的稳定性试验分析。
[0226]
3、rna完整性分析
[0227]
rna的完整性由rin值(rna integrity)进行评估,通过2100生物分析仪(g2939a,agilent)以及配套的rna 6000nano芯片(购自agilent)进行检测。
[0228]
结果见表2。实施例2制备的rna溶液的rin值分别为8.7、8.9和8.5,因此rna初始样本完整性良好,可用于后续的稳定性试验分析。与实施例2相比,对比例1制备的rna溶液的rin值稍低。因此,本发明方法所提取的rna的纯度和完整性均高于传统sds-酚仿抽提法。
[0229]
表2实施例2提取rna与对比例1提取的rna的质量对比
[0230][0231]
4、rt-pcr检测
[0232]
(1)采用primescript
tmⅱ1
st strand cdna synthesis kit(6210a,购自takara)合成第一链cdna。
[0233]
具体步骤:供试溶液4μl(rna含量为1μg)、0.5μg/μl的oligo(dt)引物1μl、10mm dntp 1μl,加不含核酸酶的水至12μl;然后65℃孵育5min,然后快速置于冰上冷却,简单离心;然后在反应体系中加入5
×
primescriptⅱbuffer 4μl、40u/μl rna酶抑制剂1μl,然后加入0.1m dtt 2μl,轻轻混匀;然后42℃孵育2min;然后,加入primescriptⅱ逆转录酶1μl,42℃孵育1h,然后70℃孵育15min使反应失活。
[0234]
(2)以第一链cdna模板,以actin基因(基因序列号:kp676600)为靶基因,进行pcr扩增。
[0235]
正向引物:5
’-
catacagtccccatctacgaag-3’;
[0236]
反向引物:5
’-
tccctcacaatttcccgc-3’。
[0237]
pcr扩增的反应体系:第一链cdna 2μl,10μm正向引物溶液1μl、10μm反向引物溶液1μl、10
×
pcr缓冲液5μl、2mm的dntp 5μl、25mm mgso
4 3μl、1u/μl dna聚合酶(购自takara)1μl,加超纯水至50μl。
[0238]
pcr扩增的反应条件:94℃3min;94℃30s、60℃30s、72℃30s,30次循环;72℃5min。
[0239]
pcr扩增产物进行1.2%琼脂糖凝胶电泳,结果见图4。实施例2提取的rna为反转录模板时,在150bp处明显可见一条清晰的特异性条带,与预期条带的分子量相同,可确定为目的产物片段。对比例1提取的rna由于混有dna,因此作为反转录模板时,产物有多条条带。因此,本方法提取的rna样本质量好,反转录效率高,可满足下游实验的要求。
[0240]
三、蛋白质质量验证
[0241]
供试溶液:实施例2制备的蛋白溶液-a1、蛋白溶液-b1或蛋白溶液-c1;对比例的步骤三制备的蛋白溶液-a4、蛋白溶液-b4或蛋白溶液-c4。
[0242]
1、蛋白浓度检测
[0243]
取供试溶液,采用bca蛋白含量测定试剂盒(23225,购自thermofisher scientific)进行检测。
[0244]
结果见表3。实施例2制备的蛋白溶液的蛋白总量稍高于对比例1制备的蛋白溶液的蛋白总量,但无明显差异。因此,本发明方法所提取的蛋白质量较高。
[0245]
表3实施例2与对比例1提取的蛋白质的质量对比
[0246][0247]
2、sds-page检测
[0248]
取供试溶液(含20μg蛋白质),采用12%的分离胶进行sds-page电泳。电压80v,电泳15分钟后加电压至150v,继续电泳50分钟。
[0249]
结果见图5。经考马斯亮蓝r250(0615-5g,购自bbi)染色后实施例2制备的蛋白溶液和对比例1制备的蛋白溶液电泳条带均很清晰,提取的蛋白没有明显差异。说明本发明方法提取的蛋白样本与商业试剂盒相比没有差异,但本发明更高效快捷,可以同时从样本中提取dna、rna和蛋白质进行综合分析。
[0250]
实施例4、同时提取独一味组织样本中的dna、rna和蛋白质
[0251]
采用实施例1制备的提取液、rna沉淀溶解液、洗涤液1、洗涤液2和蛋白裂解液。
[0252]
设置三个重复样本,分别命名为样本a、样本b和样本c。每个样本均为独一味(来源于四川省甘孜州石渠县起坞乡2村省道217路边种植)的冻存叶片组织。
[0253]
方法同实施例2。
[0254]
样本a进行上述步骤,得到的dna溶液命名为dna溶液-a,得到的rna溶液命名为rna溶液-a、得到的蛋白溶液命名为蛋白溶液-a。
[0255]
样本b进行上述步骤,得到的dna溶液命名为dna溶液-b,得到的rna溶液命名为rna溶液-b、得到的蛋白溶液命名为蛋白溶液-b。
[0256]
样本c进行上述步骤,得到的dna溶液命名为dna溶液-c,得到的rna溶液命名为rna溶液-c、得到的蛋白溶液命名为蛋白溶液-c。
[0257]
实施例5、提取效果验证
[0258]
方法基本同实施例3,本实施例中仅描述差异步骤。
[0259]
一、dna质量验证
[0260]
供试溶液:实施例4制备的dna溶液-a、dna溶液-b或dna溶液-c。
[0261]
1、dna的纯度和浓度检测
[0262]
结果见表4。实施例4制备的dna溶液的a
260
/a
280
的值均在1.8-2.0的范围内,表明
dna的纯度较高,可以用于后续的实验和分析。因此,本发明方法所提取的dna的纯度和产量均能满足下游实验的要求。
[0263]
表4实施例4提取的dna质量
[0264]
样品编号a
260
/a
280
dna浓度(ng/μl)dna总量(μg)a1.89165.516.55b1.93159.315.93c1.95161.716.17
[0265]
2、琼脂糖凝胶电泳检测
[0266]
电泳结果见图6。实施例4制备的dna溶液均在大于20kb处有一条明显dna条带,且条带完整性良好,未见rna杂质干扰以及dna条带降解。
[0267]
3、pcr扩增检测
[0268]
以供试溶液作为模板,以chs(chalcone synthase,查尔酮合酶)基因(基因序列号:kf385880)横跨外显子1-2的区域为靶序列,进行pcr扩增。
[0269]
正向引物:5
’-
cgtcgaaaatggtgaccgtgga-3’;
[0270]
反向引物:5
’-
cccgtagtcaaattaagtaacc-3’。
[0271]
pcr扩增的反应体系:供试溶液2.5μl、10μm正向引物溶液1μl、10μm反向引物溶液1μl、25μl 2
×
taq pcr mastermix(购自qiagen),加超纯水至50μl。
[0272]
pcr扩增的反应条件:94℃3min;94℃30s、60℃30s、72℃1min30s,30次循环;72℃5min。
[0273]
pcr扩增产物进行1.2%琼脂糖凝胶电泳,结果见图7。以实施例4制备的dna为模板的pcr产物均在1200bp处明显可见一条清晰的特异性条带,与预期条带的分子量相同,可确定为目的产物片段。说明本方法提取的dna样本完整性良好,可满足下游pcr实验的要求。
[0274]
二、rna质量验证
[0275]
供试溶液:实施例4制备的rna溶液-a、rna溶液-b或rna溶液-c。
[0276]
1、rna的纯度和浓度检测
[0277]
结果见表5。实施例4制备的rna溶液的a
260
/a
280
的值均稍大于2.0,表明rna的纯度较高,可以用于后续的实验和分析。因此,本发明方法所提取的rna的纯度和产量均能满足下游实验的要求。
[0278]
2、琼脂糖凝胶电泳检测
[0279]
电泳结果见图8。实施例4制备的rna溶液均显示三条明显的条带,分别对应5s、18s和28s,且未见dna的干扰。因此,本发明提取的rna样本完整性良好,未见dna干扰以及条带降解,可用于后续的稳定性试验分析。
[0280]
3、rna完整性分析
[0281]
结果见表5。实施例4制备的rna溶液的rin值分别为8.3、8.1和8.4,因此rna初始样本完整性良好,可用于后续的稳定性试验分析。
[0282]
表5实施例4提取rna的质量
[0283]
样品编号a
260
/a
280
rna浓度(ng/μl)rna总量(μg)rin值a2.03231.759.988.1b2.0622810.168.3
c2.11237.7510.078.4
[0284]
4、rt-pcr检测
[0285]
以第一链cdna模板,以actin基因(基因序列号:kf438040)为靶基因,进行pcr扩增。
[0286]
正向引物:5
’-
gcacctctcaaccccaag-3’;
[0287]
反向引物:5
’-
ctcaccccatcaccagaatc-3’。
[0288]
pcr扩增的反应体系:第一链cdna 2μl,10μm正向引物溶液1μl、10μm反向引物溶液1μl、10
×
pcr缓冲液5μl、2mm的dntp 5μl、25mm mgso4 3μl、1u/μl dna聚合酶(购自takara)1μl,加超纯水至50μl。
[0289]
pcr扩增的反应条件:94℃3min;94℃30s、60℃30s、72℃30s,30次循环;72℃5min。
[0290]
pcr扩增产物进行1.2%琼脂糖凝胶电泳,结果见图9。实施例4提取的rna为反转录模板时,在150bp处明显可见一条清晰的特异性条带,与预期条带的分子量相同,可确定为目的产物片段。因此,本方法提取的rna样本质量好,反转录效率高,可满足下游实验的要求。
[0291]
三、蛋白质质量验证
[0292]
供试溶液:实施例4制备的蛋白溶液-a、蛋白溶液-b或蛋白溶液-c。
[0293]
1、蛋白浓度检测
[0294]
结果见表6。本发明方法所提取的蛋白质量较高。
[0295]
表6实施例4提取的蛋白质的质量
[0296]
样品编号蛋白浓度(μg/μl)蛋白总量(mg)a8.41.98b7.851.87c8.752.02
[0297]
2、sds-page检测
[0298]
结果见图10。经考马斯亮蓝r250(0615-5g,购自bbi)染色后实施例4制备的蛋白溶液电泳条带均很清晰。
[0299]
以上结果说明,本发明方法可以同时从样本中提取dna、rna和蛋白质进行综合分析。
[0300]
实施例6、采用异硫氰酸胍作为变性剂、dtt作为还原剂制备提取液进行样本提取
[0301]
提取液的制备:1g ctab粉末、1g pvp k-30粉末、0.35mol异硫氰酸胍、1g tritonx-100、2.5ml tris-hcl(ph7.5、2m),加depc处理的水至50ml,使用前加入dtt并使其浓度为10mmol/l。
[0302]
rna沉淀溶解液的制备:12.5ml 4m nacl水溶液、5ml 10%(w/v)sarkosyl水溶液、500μl tris-hcl(ph8.0、1m)、100μl edta(ph8.0、0.5m),加depc处理的水至50ml。
[0303]
采用实施例1制备的洗涤液1、洗涤液2。
[0304]
样本同实施例4,方法同实施例2。
[0305]
样本a进行上述步骤,得到的dna溶液命名为dna溶液-a1,得到的rna溶液命名为rna溶液-a1。
[0306]
样本b进行上述步骤,得到的dna溶液命名为dna溶液-b1,得到的rna溶液命名为
rna溶液-b1。
[0307]
样本c进行上述步骤,得到的dna溶液命名为dna溶液-c1,得到的rna溶液命名为rna溶液-c1。
[0308]
实施例7、提取效果验证
[0309]
一、dna质量验证
[0310]
1、琼脂糖凝胶电泳检测
[0311]
供试溶液:实施例6制备的dna溶液-a1、dna溶液-b1或dna溶液-c1;实施例4制备的dna溶液-a、dna溶液-b或dna溶液-c。
[0312]
方法基本同实施例3。
[0313]
电泳结果见图11。实施例6制备的dna溶液均在大于20kb处有一条明显dna条带,且条带完整性良好,未见rna杂质干扰以及dna条带降解。与实施例4制备的dna溶液无显著差异。
[0314]
二、rna质量验证(琼脂糖凝胶电泳检测)
[0315]
供试溶液:实施例6制备的rna溶液-a1、rna溶液-b1或rna溶液-c1;实施例4制备的rna溶液-a、rna溶液-b或rna溶液-c。
[0316]
方法基本同实施例3。
[0317]
电泳结果见图12。实施例6制备的rna溶液均显示三条明显的条带,分别对应5s、18s和28s,且未见dna的干扰。虽然琼脂糖凝胶电泳结果显示实施例6制备的rna溶液亮度比实施例4稍低,暗示其产量可能稍低,但其完整性良好。因此,本发明提取的rna样本完整性良好,未见dna干扰以及条带降解,可用于后续的稳定性试验分析。
[0318]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1