葛根素-脯氨酸共晶及其制备方法与流程
[0001]
本发明属于工业结晶领域,具体涉及一种葛根素-脯氨酸共晶及其制备方法。
背景技术:
[0002]
葛根素(puerarin)为中国药典收载药物品种,化学名称为8-(β-d-吡喃葡糖 基-7-羟基-3-(4-羟苯基)-4h-1-苯并吡喃-4-酮,分子式为c
21
h
20
o9,分子结构如下 图所示:
[0003][0004]
葛根素是从中药葛根中提取的一种异黄酮类化合物,其具有降血糖、降血 脂、保护血管、抗氧化应激、抗感染及提高胰岛素敏感指数等作用,并且不良反 应少,已被临床用于治疗心脑血管疾病、癌症、帕金森病、阿尔茨海默病、糖尿 病和糖尿病并发症等疾病。然而葛根素的溶解度和生物利用度低、半衰期短,成 为限制葛根素疗效和应用的主要因素。
[0005]
在药物研发过程中,针对活性药物成分存在的缺陷,常常通过筛选药物的 固体存在形式、开发新剂型等手段加以改善。文献(药物分析杂志2004年第24 卷第2期119页)报道了葛根素的四种晶型样品的制备方法,并用pxrd、dsc、 tga和ir进行了晶型鉴定分析;中国专利cn101899041a报道了葛根素的一种 新的晶型(v型),这种新的晶型物质较原有药物具有吸收快、血药浓度高、持 续血药浓度周期平台长的特性,但从其pxrd图看,这种v晶型实际为无定型; 专利cn109662963报道了葛根素v晶型在制备预防和/或治疗糖尿肝脏损伤药 物的用途;专利cn101880275a报道了葛根素的单晶及其制备方法,单晶衍射 分析结果表明,所报道的是葛根素—水合物晶型;文献(rscadv.,2016,6,69889) 等报道了将葛根素分散在聚氰基丙烯酸丁酯中制备成纳米颗粒,以改善其生物利 用度。
[0006]
共晶作为一种新型的固体存在形态,将药物活性成分(active pharmaceuticalingredients,api)与合适的共晶形成物(co-crystal former,ccf)通过氢键自组 装,或者通过带有饱和性和方向性的非共价键(如芳烃或苯环的范德华力,π-π 共轭作用和卤键)组装形成的一种新型结构,药物与共晶形成物间存在稳定的化 学计量比。通过药物共晶技术来制备药物共晶,可获得更加多样化的药物固体形 态,同时药物共晶之所以对制药工业有很大的吸引力在于它提供了一种不需要破 坏和产生共价键就能够达到修饰药物活性成分(api)的物理或化学性质的机会。 在保留药物本身的药理性质的同时,达到了修饰药物的理化性质的目的,这一点 为药物共晶在制药工业方面的应用提供了更为广阔的发展空间。在不改变药物结 构及本身的药理性质的情况下,形成的新晶体可以提高药物的溶解性、溶出度、 生物利用度、稳定性,降低引湿性,改善机械性能等。因此,获得更多具有新颖、 实用和创造性的药物共晶具有重要的现实意义,特别是一些水不溶性药物。近几 年来,药物共
晶研究越来越受到人们的关注。现阶段,国外对药物共晶的研究开 始逐渐增多并深入;而国内对其研究还相对较少。
技术实现要素:
[0007]
本发明的目的在于提供一种改进的葛根素的固体存在形式,特别是葛根素的 共晶,其在物理上对温度胁迫保持稳定;另一个目的在于提供一种改进的葛根素 的固态形式,特别是葛根素的共晶,其在化学上对光降解保持稳定。其特征在于 改善的溶解度/溶出度和/或特征在于改善的粉末特性,例如流动性、堆密度和可 压缩性。
[0008]
本发明通过发现葛根素与l-脯氨酸的药物共晶解决了上述确定目标的一个 或多个,制备得到了葛根素和l-脯氨酸的两种共晶。葛根素共晶的形成提高了 原料药葛根素的溶解性、稳定性和生物利用度,并为葛根素口服制剂的研发提供 了一条途径。同时,本发明还提供了葛根素-l-脯氨酸共晶的制备方法。
[0009]
为了实现本发明的技术目的,本发明的具体技术方案如下:
[0010]
本发明公开了一种葛根素-脯氨酸共晶ⅰ,以葛根素为药物活性成分,以l
-ꢀ
脯氨酸为共晶形成物,由一个葛根素分子、一个l-脯氨酸分子组成葛根素-脯氨 酸共晶ⅰ的基本结构单元,葛根素-脯氨酸共晶ⅰ属单斜晶系,p 21空间群,晶 胞参数为:轴长轴角 β=97.7830(10)
°
,z=2。
[0011]
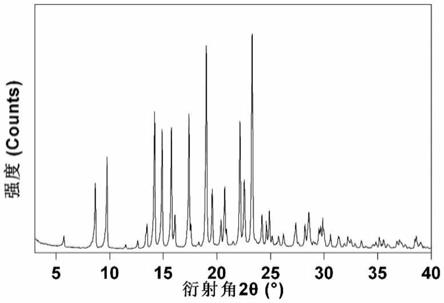
作为进一步地改进,本发明所述的葛根素-脯氨酸共晶ⅰ具有如图1所示的 x射线衍射图谱,其以衍射角2θ
±
0.1
°
表示的x-射线衍射包含5.7,8.7,9.7, 13.5,14.2,14.9,15.8,17.4,19.0,19.6,20.7,22.1,22.6,23.3的处 的特征峰。
[0012]
本发明还公开了一种葛根素-脯氨酸共晶ⅰ的制备方法,具体制备步骤如下:
[0013]
1)、将摩尔比1:1的葛根素和l-脯氨酸,加入到醇和水的混合溶剂中, 加热回流溶解,反应体系中溶质的重量mg和溶液的体积ml的比为100~310;
[0014]
2)、溶解的温度在65~85℃;
[0015]
3)、溶解澄清后,搅拌5~10分钟,即有白色固体体析出,继续搅拌或超 声0.5至2小时,缓慢冷却至35~50℃,即得葛根素-脯氨酸共晶ⅰ。
[0016]
作为进一步地改进,本发明所述的醇为甲醇、乙醇、丙醇、丁醇或戊醇,醇 和水的体积比为10:1~8:2。
[0017]
作为进一步地改进,本发明所述的醇为乙醇。
[0018]
本发明还公开了一种葛根素-脯氨酸共晶ⅱ,以葛根素为药物活性成分,以 l-脯氨酸为共晶形成物,由两个葛根素分子、一个l-脯氨酸分子组成、二个水 分子和0~1个乙醇分子组成葛根素-脯氨酸共晶ⅱ的基本结构单元,所述的葛根 素-脯氨酸共晶ⅱ属三斜晶系,p1(1
#
)的空间群,其晶胞参数:轴长轴长轴角α=81.7330(10)
°
,β=89.7330 (10)
°
,γ=87.4870(10)
°
,z=1。
[0019]
作为进一步地改进,本发明所述的葛根素-脯氨酸共晶ⅱ具有如图7所示的 x射线衍射图谱,其以衍射角2θ
±
0.1
°
表示的x-射线衍射包含5.7,7.4,8.6, 9.9,11.4,12.6,14.0,15.2,16.2,16.8,17.5,18.1,18.7,20.4,20.9, 21.9,22.7,23.0,24.9,25.4的处的
特征峰。
[0020]
本发明还公开了一种葛根素-脯氨酸共晶ⅱ的制备方法,具体制备步骤如下:
[0021]
1)、将摩尔比为2:1的葛根素和l-脯氨酸加入溶剂中,反应体系中溶质的 重量mg和溶液的体积ml的比为100~260;
[0022]
2)、60~80℃加热回流,搅拌,完全溶解后再继续搅拌0.5小时~3小时, 逐渐有固体析出;
[0023]
3)、停止搅拌,降温至0~25℃,保温析晶0.5~2小时让其析出完全,析出 物即为所述的葛根素-l-脯氨酸共晶ⅱ。
[0024]
作为进一步地改进,本发明所述的溶剂为乙醇和水的混合溶剂,乙醇与水 的体积比为10:1~7:3。
[0025]
本发明相对于现有技术的有益效果如下:
[0026]
1.本发明针对葛根素水溶性差的缺点,采用共晶技术,改善其水溶性。溶 出速率和溶解度实验显示,葛根素-l-脯氨酸的两种共晶在37℃下,ph1.2酸性 溶液、ph6.8磷酸缓冲溶液和水中的溶解度和溶出速率均优于葛根素,共晶ⅰ在 ph1.2酸性溶液、ph6.8磷酸缓冲溶液和水中的平衡溶解度(24小时)分别为葛 根素的2.6倍、1.8倍和2.3倍;共晶ⅱ在ph1.2酸性溶液、ph6.8磷酸缓冲溶液 和水中的平衡溶解度(24小时)分别为葛根素的2.3倍、1.9倍和2.3倍。在ph1.2 酸性溶液中,共晶i和共晶ii的1小时溶出速率是葛根素的2.5倍和3.8倍;在 ph6.8磷酸缓冲溶液中,共晶i和共晶ii的1小时溶出速率是葛根素的1.8倍和 4.4倍;在水中,共晶i和共晶ii的1小时溶出速率是葛根素的1.3倍和3.1倍。 (详见表3)。暗示着在生物利用度方面将会有明显的改善,溶解后可以制成更 多的剂型,如口服制剂等,丰富了葛根素的剂型,提高了该药物的应用范围。
[0027]
表3共晶i和共晶ii在ph1.2酸性溶液、ph6.8磷酸缓冲溶液和水中的溶解 度
[0028][0029]
2.本发明采用了葛根素与脯氨酸组装,生成新的葛根素-l-脯氨酸共晶,并 对其结构进行分析和表征。众所周知,氨基酸除了生成能量以外,还有多种生理 功效,如促进蛋白合成、保护肝功能、保护心肌功能以及提高免疫、消除疲劳的 功能等等,改进后的药物不仅具有降血糖、降血脂、保护血管、抗氧化应激、抗 感染及提高胰岛素敏感指数等作用,还增加脯氨酸具有的功能。
[0030]
3.本发明的共晶,制备方法简便易行,使用绿色溶剂,条件温和,适合工 业化生
产。
附图说明
[0031]
图1是葛根素-l-脯氨酸共晶i的pxrd图;
[0032]
图2是葛根素-l-脯氨酸共晶i的晶体结构图(ortep图);
[0033]
图3是葛根素-l-脯氨酸共晶i的氢键连接图;
[0034]
图4是葛根素-l-脯氨酸共晶i的热分析(tg/dsc)图;
[0035]
图5是葛根素-l-脯氨酸共晶i的raman谱图;
[0036]
图6是葛根素-l-脯氨酸共晶i的ir谱图;
[0037]
图7是葛根素-l-脯氨酸共晶ii的pxrd图;
[0038]
图8是葛根素-l-脯氨酸共晶ii的晶体结构图(ortep图);
[0039]
图9是葛根素-l-脯氨酸共晶ii的氢键连接图;
[0040]
图10是葛根素-l-脯氨酸共晶ii的堆叠图;
[0041]
图11是葛根素-l-脯氨酸共晶ii的热分析(tg/dsc)图;
[0042]
图12是葛根素-l-脯氨酸共晶ii的raman谱图;
[0043]
图13是葛根素-l-脯氨酸共晶ii的ir谱图;
[0044]
图14是葛根素、共晶i和共晶ii在ph1.2酸性溶液中溶出速率图;
[0045]
图15是葛根素、共晶i和共晶ii在ph6.8缓冲溶液中溶出速率图;
[0046]
图16是葛根素、共晶i和共晶ii在水中溶出速率图;
[0047]
图17是分别是葛根素、共晶i和共晶ii在ph1.2酸性溶液、ph6.8缓冲溶液、 水中的平衡溶解度(平衡24小时)。
具体实施方式
[0048]
本发明涉及葛根素的物理化学性质和/或药物性质的改善。在此公开了葛根素 的两种新型共晶,包括1:1葛根素-l-脯氨酸共晶i、2:1葛根素-l-脯氨酸共晶ii。
[0049]
一种葛根素-l-脯氨酸共晶i,以葛根素为药物活性成份,l-脯氨酸为共晶形 成物,在170k下用单晶x射线衍射分析并确定了晶体结构。一个葛根素分子, 一个l-脯氨酸分子组成葛根素药物共晶i的基本结构单元,其中葛根素分子中的 羟基既作为氢键受体,又作为氢键给体,分别与l-脯氨酸中的氨基和羰基形成 两种不同的分子间氢键,所形成的药物共晶属单斜晶系,p21(4
#
)的空间群, 其晶胞参数:轴长轴 角β=97.7830(10)
°
,z=2。
[0050]
表1 共晶i的晶体数据表
[0051][0052]
葛根素-脯氨酸共晶ⅰ具有如图1所示的x射线衍射图谱,其以角2θ
±
0.1
°ꢀ
表示的x-射线衍射包含5.7,8.7,9.7,13.5,14.2,14.9,15.8,17.4,19.0,19.6, 20.7,22.1,22.6,23.3的处的特征峰。
[0053]
本发明药物共晶i的热重谱图(tg)显示,在室温至150℃范围内没有失 重,表明结构中不含溶剂(包括水);其差热谱图(dsc)显示,在224.4℃(峰 顶值)处有一个吸热峰,为该药物共晶的熔融分解峰。
[0054]
本发明药物共晶i的红外谱图(ir)显示,在3389cm-1
,2923cm-1
,1632cm-1
, 1515cm-1
,1446cm-1
,1268cm-1
,1214cm-1
,1176cm-1
,1085cm-1
,1061cm-1
处有特征吸收峰。
[0055]
本发明还提供了一种葛根素-l-脯氨酸共晶i的制备方法,具体步骤如下: 将摩尔比为1:1的葛根素和l-脯氨酸加入到醇和水的混合溶剂,加热至65~85℃, 搅拌或超声直到完全溶解后,继续搅拌或超声0.5至2小时,缓慢冷却至35~50℃, 静置或搅拌析晶,得到葛根素-l-脯氨酸共晶i;醇为甲醇、乙醇、丙醇、丁醇或 戊醇,醇优选为乙醇;混合溶剂中醇的体积ml和水的体积ml比为10:1~8:2, 反应溶液体系中溶质含量为100~310mg/ml。
[0056]
本发明还公开了一种葛根素-l-脯氨酸共晶ii,以葛根素为药物活性成份, l-脯氨酸为共晶形成物。二个葛根素分子,一个l-脯氨酸分子,二个水分子和 0~1个乙醇分子组成葛根素药物共晶ii的基本结构单元;其中葛根素分子中的羟 基既作为氢键给体,与l-脯氨酸中的羰基形成氢键;所形成的药物共晶属三斜晶 系,p1(1
#
)的空间群,其晶胞参数:轴长轴长轴角α=81.7330(10)
°
,β=89.7330(10)
°
,γ=87.4870(10)
ꢀ°
,z=1。
[0057]
表2 共晶i的晶体数据表
[0058][0059]
葛根素-脯氨酸共晶ⅱ具有如图7所示的x射线衍射图谱,其以角2θ
±
0.1
°ꢀ
表示的x-射线衍射包含5.7,7.4,8.6,9.9,11.4,12.6,14.0,15.2,16.2,16.8, 17.5,18.1,18.7,20.4,20.9,21.9,22.7,23.0,24.9,25.4的处的特征峰。
[0060]
本发明的葛根素-l-脯氨酸共晶ii的热重谱图(tg)显示,在室温至150℃ 范围内有3.8~7.8%的失重;其差热谱图(dsc)显示,在峰顶值为112.7℃处有 一个脱去溶剂引起的吸热峰,其熔融分解温度(峰顶值)为195.6℃。药物共晶 的红外谱图(ir)显示,在3361cm-1
,3084cm-1
,3018cm-1
,2924cm-1
,2855cm-1
, 2736cm-1
,2673cm-1
,2607cm-1
,1646cm-1
,1605cm-1
,1515cm-1
处有特征吸收 峰。
[0061]
本发明还提供了一种葛根素-l-脯氨酸共晶ii的制备方法,具体步骤如下:
[0062]
将摩尔比为2:1的葛根素和l-脯氨酸加入到乙醇和水的混合溶剂,加热至 60~80℃,搅拌或超声直到完全溶解后,继续搅拌或超声0.5至3小时,缓慢冷 却至0~25℃,静止析晶,得到葛根素-l-脯氨酸共晶ii;混合溶剂中乙醇的体积 ml和水的体积ml比为10:1~7:3,反应溶液体系中溶质含量为100~260mg/ml。
[0063]
本发明中表征葛根素共晶的结构及性能的仪器如下:
[0064]
单晶x射线衍射仪:单晶衍射数据用brukerd8ventureiμs3.0单晶衍射仪 (cukα,管电压50kv,管电流1.1ma,检测器photoniidetector) 在297(2)k温度下采集。晶体结构解析采用直接法,随后采用数轮差值fourier 合成法确定了全部非氢原子的坐标,继而用全矩阵最小二乘法对所有非氢原子进 行各向异性温度因子修正。氢原子的位置通过fourier合成法确定,但采用理论 加氢方式加氢,并采用riding模式进行修正。
[0065]
综合热分析仪:美国ta公司,型号为sdtq600,吹扫气:氮气100ml/min, 升温速度10℃/min,温度范围:室温~400℃。
[0066]
差示扫描量热仪:美国ta公司,型号为dscq100,吹扫气:氮气50ml/min, 升温速度10℃/min,温度范围:室温~300℃。
[0067]
拉曼光谱(raman):日本堀场horiba公司,型号为labramhrevolution, 拉曼采集
激光波长为633nm的he-ne激光器,并用10
×
显微镜收集背散射光.光 栅为600gr/mm,采集时间10s,光谱范围200-4000cm-1
,仪器的光谱分辨率为 2cm-1
,激光输出能量为10mw。
[0068]
紫外吸收光谱(uv):采用dynamicahalodb-20紫外分光光度计,将样品 配制成一定浓度溶液,并用同批溶剂作为空白对照,采用1cm吸收池在 200~400nm范围内测定。仪器校正与检定按中国药典2015版附录进行校正。
[0069]
下面通过附图和实施例进一步对本发明中所述方法进行描述,但本发明保护 范围不受实施例限制,以下实施例只是描述性的,而非限定性的,不能以限定本 发明的保护范围。
[0070]
实施例1葛根素-l-脯氨酸共晶i的制备
[0071]
精密称取葛根素0.832g(0.5mmol)和脯氨酸0.230g(0.5mmol)至25ml圆 底烧瓶中,加80%(体积比)乙醇6ml放入水浴锅中边搅拌边加热到85℃,溶 清后继续搅拌,约5~10分钟后即析出固体,继续搅拌1小时,然后边搅拌边降 温到50℃,保温静置析晶0.5小时,抽滤,50℃干燥2小时得产物。
[0072]
实施例2葛根素-l-脯氨酸共晶i的制备
[0073]
精密称取葛根素0.832g(0.5mmol)和脯氨酸0.230g(0.5mmol)至25ml圆 底烧瓶中,加90%(体积比)乙醇7ml放入水浴锅中边搅拌边加热到80℃,溶 清后搅拌约5~10分钟后即析出固体,继续搅拌时间为1~2小时,边搅拌边自然 降温至45℃,停止搅拌,静置析晶约0.5小时,抽滤,40℃干燥4小时得产物。
[0074]
实施例3葛根素-l-脯氨酸共晶i的制备
[0075]
精密称取葛根素0.832g(0.5mmol)和脯氨酸0.230g(0.5mmol)至25ml圆 底烧瓶中,加90%(体积比)甲醇5ml,加热回流(约65℃)搅拌,溶清后继 续搅拌约10分钟即析出固体,继续搅拌1小时,自然降温至35℃,停止搅拌, 35℃保温析晶1小时,抽滤,50℃干燥2小时。
[0076]
实施例4葛根素-l-脯氨酸共晶i的制备
[0077]
精密称取葛根素0.832g(0.5mmol)和脯氨酸0.230g(0.5mmol)至25ml圆 底烧瓶中,加入85%(体积比)异丙醇10ml,加热回流(约80-85℃)搅拌, 溶清后搅拌10~15分钟即析出固体,继续搅拌1小时,自然降温至50℃,停止 搅拌,50℃保温静置析晶1小时,抽滤,45℃干燥3小时。
[0078]
实施例5葛根素-l-脯氨酸共晶i的制备
[0079]
精密称取葛根素1.664g(1mmol)和脯氨酸0.460g(1mmol)至50ml圆底 烧瓶中,加入90%(体积比)乙醇20ml,加热回流(约80℃)搅拌,溶清后搅 拌约5~10分钟即析出固体,继续搅拌2小时,然后自然降温至40℃,停止搅拌, 40℃保温静置析晶0.5小时,抽滤,50℃干燥2小时。
[0080]
实施例6葛根素-l-脯氨酸共晶i的制备
[0081]
精密称取葛根素16.64g(10mmol)和脯氨酸4.60g(10mmol)至100ml圆 底烧瓶中,加入65ml 80%(体积比)乙醇,搅拌加热回流(约85℃),溶清后 搅拌约5~10分钟即析出大量白色固体,继续搅拌3小时,然后自然降温至35℃, 停止搅拌,35℃静置析晶1小时,抽滤,50℃干燥2小时。
[0082]
对共晶i产物进行化学性质表征:
[0083]
图1和表4为葛根素-l-脯氨酸共晶i的实验pxrd图和特征衍射谱线表。这 些峰的全部列表或其子集可以表征所述共晶。例如,可以通过选自在2θ
±
0.1
°
: 5.7,8.7,9.7,13.5,14.2,14.9,15.8,16.1,17.4,19.0,19.6,20.7,22.1, 22.6,23.3下的峰至少5个峰以及通过基本上类似于图1的pxrd图来表征共晶。
[0084]
表4 共晶i的特征衍射谱线表
[0085]
[0086][0087]
葛根素-l-脯氨酸共晶i的scxrd分析结果表明,其最小不对称单元中含有 一个葛根素分子,一个l-脯氨酸分子,其中葛根素分子中的羟基既作为氢键受 体,又作为氢键给体,分别与l-脯氨酸中的氨基和羰基形成两种不同的分子间 氢键。图2和图3分别为葛根素-l-脯氨酸共晶i的晶体结构图(椭球概率50%) 和氢键连接图。
[0088]
葛根素-l-脯氨酸共晶i的热重分析图(tg,图4)显示,在室温~150℃范 围内没有失重,表明结构中不含溶剂,与单晶衍射分析结果一致。差示扫描量热 法(dsc,图4)图显示,葛根素-l-脯氨酸共晶i在熔融分解温度为224.4℃(峰 顶值)。
[0089]
葛根素-l-脯氨酸共晶i的raman谱图(图5),显示了以下的特征峰:3376 cm-1
、3064cm-1
、3046cm-1
、3015cm-1
、2985cm-1
、2924cm-1
、2904cm-1
、2876 cm-1
、2850cm-1
。
[0090]
葛根素-l-脯氨酸共晶i的红外谱图(图6),显示了以下的特征峰ir(kbr): 3389(br)cm-1
,3248(sh)cm-1
,2923(m)cm-1
,1632(s)cm-1
,1592(s)cm-1
,1515(s) cm-1
,1446(s)cm-1
,1398(s)cm-1
,1268(s)cm-1
,1214(s)cm-1
,1176(m)cm-1
,1085(s) cm-1
,1061(s)cm-1
,1014(s)cm-1
,890(m)cm-1
,837(m)cm-1
,797(m)cm-1
,632(m) cm-1
,602(m)cm-1
,546(m)cm-1
。
[0091]
实施例7葛根素-l-脯氨酸共晶ii的制备
[0092]
精密称取葛根素1.644g(1mmol)和脯氨酸0.230g(0.5mmol)至50ml圆底 烧瓶中,加入18ml 90%(体积比)乙醇,搅拌加热回流(80℃),溶清后继续 搅拌2小时,逐渐有固体析出,自然降温至室温(20℃-25℃),停止搅拌,静置 析晶1小时,抽滤,40℃干燥4小时。
[0093]
实施例8葛根素-l-脯氨酸共晶ii的制备
[0094]
精密称取葛根素1.644g(1mmol)和脯氨酸0.230g(0.5mmol)至50ml圆底 烧瓶中,加入15ml 80%(体积比)乙醇,边搅拌边加热至60℃直至溶清,继 续搅拌2小时,逐渐有固体析出,停止搅拌,自然降温到室温(20℃-25℃),抽 滤,40℃干燥4小时。
[0095]
实施例9葛根素-l-脯氨酸共晶ii的制备
[0096]
精密称取葛根素1.644g(1mmol)和脯氨酸0.230g(0.5mmol)至25ml圆底 烧瓶中,加入70%(体积比)乙醇7ml,边搅拌边加热到70℃,溶清后继续搅 拌2小时,逐渐有固体析出,自然降温,降至室温后放入冰水浴中(0℃-5℃), 继续静置析晶1小时,抽滤工,40℃干燥4小时得产物。
[0097]
实施例10葛根素-l-脯氨酸共晶ii的制备
[0098]
精密称取葛根素16.64g(10mmol)和脯氨酸2.30g(5mmol)至250ml圆底 烧瓶中,加入180ml 90%(体积比)的乙醇,边搅拌边加热至75℃直至溶清, 溶清后继续搅拌2小时,逐渐有固体析出,自然降温至室温(20℃-25℃),停止 搅拌,静置析晶1~2小时,抽滤,40℃干燥4小时。
[0099]
对葛根素-l-脯氨酸共晶ii进行化学表征:
[0100]
图7和表3为葛根素-l-脯氨酸共晶ii的实验pxrd图和特征衍射谱线表。 这些峰的全部列表或其子集可以表征所述共晶。例如,可以通过选自在2θ(
±
0.1
°
) 5.7,7.4,8.6,11.4,12.6,14.0,15.2,16.2,16.8,17.5,18.1,18.7,20.4, 20.9,21.9,22.7,23.0,24.9,25.4中的峰至少5个峰以及通过基本上类似于图 1的pxrd图来表征共晶。
[0101]
表5 共晶ii的特征衍射谱线表
[0102]
[0103][0104]
葛根素-l-脯氨酸共晶ii的scxrd的结果表明,其最小不对称单元中含有二 个葛根素分子,一个l-脯氨酸分子,一个乙醇分子和二个水分子,葛根素中的 羟基与脯氨酸中的羰基形成氢键,水分子中的氧原子和乙醇分子中的羟基作为
ꢀ“
桥原子”通过o-h
···
o,n-h
···
o和o-h
···
n氢键作用将葛根素分子和脯氨酸分子 连接,形成了丰富的氢键网络。图8和图9分别为葛根素-l-脯氨酸共晶ii的晶 体结构图(椭球概率50%)和氢键连接图。乙醇分子与葛根素分子和脯氨酸分子 间的氢键作用较弱,室温下即易从晶格中部分或全部逸出,但由于乙醇分子处中 共晶分子组成的层状结构的间隙中(图10),乙醇分子的部分或全部逸出并不 影响共晶ⅱ的晶体结构及其稳定性。
[0105]
葛根素-l-脯氨酸共晶ii的热重分析图(tg,图11)显示,在室温至150℃ 范围内的失重率为5.8%,表明结构中含有溶剂,与单晶衍射分析结果基本一致。 差示扫描量热法(dsc,图11)图显示,在峰顶值(112.7℃)处有一个脱去溶 剂引起的吸热峰,共晶ii的熔融
分解为211.3℃(峰顶值)。
[0106]
葛根素-l-脯氨酸共晶ii的raman谱图(图12),显示了以下的特征峰(cm-1
): 3395、3227、3066、3020、3001、2987、2943、2923、2892。
[0107]
葛根素-l-脯氨酸共晶ii的红外谱图(图13),显示了以下的特征峰ir(kbr) (cm-1
):3361(br),3266(sh),3084(m),3018(m),2924(w),2855(w),2736(w), 2673(w),2607(w),1646(s),1605(s),1515(s),1445(s),1387(s),1358(s),1339(s), 1258(s),1280(s),1180(s),1085(s),1043(s),1015(m),989(s),943(s),888(w), 834(m),795(w),773(w),737(w),657(w),630(m),545(m),514(m),490(w), 440(w)cm-1
处有特征峰。
[0108]
实施例11测定葛根素-l-脯氨酸共晶i和葛根素-l-脯氨酸共晶ii在ph1.2 酸性溶液中的溶出速率,并与葛根素进行对比。
[0109]
分别取适量葛根素样品、共晶i样品和共晶ii样品加入ph1.2酸性溶液中, 置水浴锅中,以温度37℃,转速150rpm缓慢搅拌,在5、10、15、20、25、30、 40、50、60、80、100、120min时取样3ml过0.24μm的微孔滤膜至离心管中, 每次取样完成后加等温等量的溶出空白介质。为使测定得到的吸光度值在规定范 围内,取样得到的澄清溶液用ph1.2酸性溶液稀释一定的倍数,在最大吸收波长 处测定所制备溶液的吸光度。每个样品平行测定3次。用标准曲线法计算其对应 的浓度,绘制出对应的溶出曲线。如图14显示。
[0110]
实施例12测定葛根素-l-脯氨酸共晶i和葛根素-l-脯氨酸共晶ii在ph6.8 磷酸缓冲溶液中的溶出速率,并与葛根素进行对比。
[0111]
分别取适量葛根素样品、共晶i样品和共晶ii样品加入ph6.8磷酸缓冲溶液 中,置水浴锅中,以温度37℃,转速150rpm缓慢搅拌,在5、10、15、20、25、 30、40、50、60、80、100、120min时取样3ml过0.24μm的微孔滤膜至离心管 中,每次取样完成后加等温等量的溶出空白介质。为使测定得到的吸光度值在规 定范围内,取样得到的澄清溶液用ph6.8磷酸缓冲溶液稀释一定的倍数,在最大 吸收波长处测定所制备溶液的吸光度。每个样品平行测定3次。用标准曲线法计 算其对应的浓度,绘制出对应的溶出曲线。如图15显示。
[0112]
实施例13测定葛根素-l-脯氨酸共晶i和葛根素-l-脯氨酸共晶ii在水中的 溶出速率,并与葛根素进行对比。
[0113]
分别取适量葛根素样品、共晶i样品和共晶ii样品加入去离子水中,置水浴 锅中,以温度37℃,转速150rpm缓慢搅拌,在5、10、15、20、25、30、40、 50、60、80、100、120min时取样3ml过0.24μm的微孔滤膜至离心管中,每次 取样完成后加等温等量的溶出空白介质。为使测定得到的吸光度值在规定范围 内,取样得到的澄清溶液用去离子水稀释一定的倍数,在最大吸收波长处测定所 制备溶液的吸光度。每个样品平行测定3次。用标准曲线法计算其对应的浓度, 绘制出对应的溶出曲线。如图16显示。
[0114]
实施例14测定葛根素-l-脯氨酸共晶i和葛根素-l-脯氨酸共晶ii在ph1.2 酸性溶液、ph6.8磷酸缓冲溶液、水中的24小时平衡溶解度,并与葛根素进行 对比。
[0115]
称取适量的葛根素样品、共晶i样品和共晶ii样品,加入适量的ph1.2酸性 溶液、ph6.8磷酸缓冲溶液、去离子水,于37
±
0.5℃恒温下以500rpm转速搅拌 24小时,达到溶解平衡后,用0.45μm的过滤器过滤,弃去初滤液,取续滤液在 最大吸收波长处测定吸光度值,用标准曲线法计算其对应的浓度,得到平衡溶解 度。如图17所示。
[0116]
最后,还需要注意的是,以上列举的仅是本发明的几个具体实施例。显然, 本发明
不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发 明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1