一种合成恩替卡韦中间体的方法与流程
[0001]
本发明属于药物合成技术领域,具体涉及一种恩替卡韦中间体4-甲氧苯基二苯基氯甲烷的合成方法。
背景技术:
[0002]
恩替卡韦(式a),化学名为2-氨基-1,9-二氢-9-[(1s,3r,4s)-4-羟基-3-(羟甲基)-2-亚甲基环戊基]-6h-嘌呤-6-酮,英文名称为entecavir,商品名为baraclude,系百时美施贵宝公司研发的一种碳环核苷类物质,具有极强的抗hbv能力。该药于2005年3月获得美国fda批准上市,2006年在中国上市。大量体外试验表明,恩替卡韦只对hbv具有抗病毒活性,而对其他dna病毒和hiv无效,因而具有高度的特异性。恩替卡韦抑制乙肝病毒的速度快且不反弹,口服吸收度好,生物利用度高,半衰期长,作用持久,耐药性低。
[0003]
恩替卡韦的结构式为:
[0004]
文献报道的恩替卡韦的合成方法较多,其中百时美施贵宝公司公布的原研工艺由于原料和试剂在国内简单易得,是目前已经工业化的工艺。该工艺以环戊二烯为原料,与金属钠反应得茂钠后经烷基化、不对称硼氢化-氧化反应得到取代环戊烯中间体,该中间体经环氧化、上保护基得环氧中间体,该环氧中间体与o-6-苄基鸟嘌呤经双分子亲核取代反应,得到环氧开环的醇中间体,该醇中间体再与4-甲氧苯基二苯基氯甲烷发生n-烷基化反应,得到苄胺中间体,该苄胺中间体再经dess-martin氧化和亚甲基化反应,最后脱保护得到恩替卡韦。因此式(i)化合物4-甲氧苯基二苯基氯甲烷是合成恩替卡韦的重要中间体,对于恩替卡韦的合成具有非常重要的意义。文献报道式(i)化合物4-甲氧苯基二苯基氯甲烷的合成主要有以下几种方法:
[0005]
第一种方法以对溴苯甲醚为原料,先制备溴化镁格氏试剂,然后与二苯甲酮发生格氏反应,制备苄醇中间体,再以乙酰氯、二氯亚砜或氯化氢为氯代试剂发生羟基的氯代反应,合成式(i)化合物。
[0006]
合成路线为:
[0007]
因该工艺第一步和第二步需制备格氏试剂和发生格氏反应,对水分要求非常高;第三步氯代反应中使用乙酰氯等氯代试剂会产生大量盐酸,除腐蚀设备外,还会对环境造成污染;另外,若用乙酰氯作为氯代试剂,还会生成较难处理的羟基乙酰化的杂质。因此,该工艺不易工业化方便操作。
[0008]
第二种方法以4-羟基三苯基甲醇为主要原料,经甲基化、氯代两步反应,合成式(i)化合物。
[0009]
合成路线为:
[0010]
此工艺在甲基化反应中,容易发生酚羟基和醇羟基同时甲基化的杂质,这个杂质与式(i)化合物结构较相似,很难除去;甲基化中选择硫酸二甲酯为甲基化试剂,该试剂毒性较大;另外,原料4-羟基三苯基甲醇价高、不易得,不利于工业化生产。
[0011]
第三种方法以二苯基二氯甲烷、苯甲醚为原料,经傅-克反应,一步合成式(i)化合物。
[0012]
合成路线为:
[0013]
此工艺比较简单,但该傅-克反应由于使用常用的催化活性很强的alcl3为催化剂,且反应在均相中进行,除了易在甲氧基对位发生傅-克反应生成式(i)化合物外,还会在甲氧基邻位发生傅-克反应产生邻位傅-克反应产物,以及产生二苯基二氯甲烷自身苯环发生的傅-克反应的产物等杂质。多种杂质极性相近,与主产物很难分离除去,另外,傅-克反应后用水洗去除alcl3催化剂的过程中,会引起式(i)化合物分子中氯原子的水解,因此,水处理干燥后还需用乙酰氯或二氯亚砜等氯代试剂进行氯代;还有,反应起始原料二苯基二氯甲烷也不易得到。因此,该工艺也不适合制备高纯度的式(i)化合物。
技术实现要素:
[0014]
本发明所要解决的技术问题是克服现有合成式(i)化合物4-甲氧苯基二苯基氯甲
烷报道的技术中原料不易得、易产生难以控制的杂质、产品纯度低、生产成本高、操作复杂、工艺安全性差、污染环境等不利于工业化生产的缺陷,提供一种有效的制备式(i)化合物4-甲氧苯基二苯基氯甲烷的方法,该方法原料价廉、易得、不易产生杂质、产品纯度高、反应条件温和、步骤简单、催化剂可重复使用、生产成本低,适合工业化生产。
[0015]
本发明的技术方案概述如下:
[0016]
步骤(1),将式(ii)化合物四氯化碳和苯在催化剂1作用下发生傅-克烷基化反应,得到式(iii)化合物二苯基二氯甲烷;步骤(2),式(iii)化合物和苯甲醚在催化剂2作用下发生傅-克烷基化反应,即制得式(i)化合物4-甲氧苯基二苯基氯甲烷。
[0017]
合成路线为:
[0018]
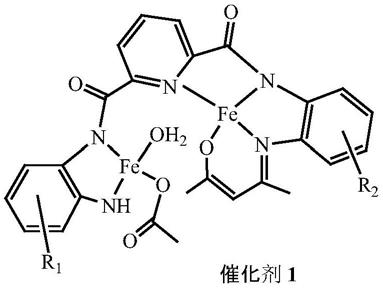
所述的步骤(1)中的催化剂1为西佛碱双核铁配合物,结构为:
[0019]
催化剂1结构中r1和r2分别为氢原子、c1~c4的烷基、c1~c4的芳烷基、卤素等。
[0020]
催化剂1制备比较简单,用常规的实验方法较容易制备。
[0021]
催化剂1催化效率较高,其用量只为四氯化碳物质的量的8~15%。
[0022]
所述的步骤(1)中的反应温度为50~75℃。
[0023]
所述的步骤(2)中的催化剂2为铁杂多酸负载sba-15介孔分子筛,催化剂的通式为:fe-pom-octyl-si-nh
3-sba-15,pom为杂多酸,fe-pom的结构为pw
11
feo
40n-;-octyl-si为正辛基三甲基硅烷;sba-15为介孔氧化硅分子筛;催化剂孔径在5-7nm之间。
[0024]
催化剂2制备也较简单,用常规的实验方法很容易制备。反应后经过滤晾干后可直接循环使用5次以上,每重复使用1次,收率约下降1%。
[0025]
所述的催化剂2催化效率高,催化剂2的质量与苯甲醚物质的量的比为3~8g:1mol。
[0026]
在催化剂2作用下,步骤2的反应在非均相体系中进行,收率高,反应选择性好,基本只在甲氧基对位发生傅-克反应生成式(i)化合物,基本不产生甲氧基邻位的傅-克反应杂质及二苯基二氯甲烷自身苯环的傅-克反应杂质。
[0027]
所述的步骤(2)中的反应温度为30~60℃。
[0028]
本工艺的优点:副产物少,产品质量易控制,产物纯度高,所用的试剂价廉、易得,
操作简单,总收率也较高。
具体实施方式
[0029]
下面结合实施具体实施例,进一步说明本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
[0030]
实施例中所用的原料或试剂除特别说明之外,均市售可得。
[0031]
实施例1式(iii)化合物二苯基二氯甲烷的制备
[0032]
将100mmol四氯化碳、10mmol催化剂1(r1和r2都为氢原子)、220mmol苯依次加入反应瓶中,室温搅拌均匀,通氮气,于65℃下搅拌反应10h,停止加热,冷却至室温,向反应混合物中加入5ml水,搅拌后分出有机层,无水硫酸钠干燥,减压蒸去苯和未反应的四氯化碳,得式(iii)化合物,收率87%。该粗产品不用纯化,直接进行下一步反应。
[0033]
实施例2式(iii)化合物二苯基二氯甲烷的制备
[0034]
将100mmol四氯化碳、10mmol催化剂1(r1和r2都为2-甲基)、220mmol苯依次加入反应瓶中,室温搅拌均匀,通氮气,于70℃下搅拌反应12h,停止加热,冷却至室温,向反应混合物中加入5ml水,搅拌后分出有机层,无水硫酸钠干燥,减压蒸去苯和未反应的四氯化碳,得式(iii)化合物,收率90%。该粗产品不用纯化,直接进行下一步反应。
[0035]
实施例3式(iii)化合物二苯基二氯甲烷的制备
[0036]
将100mmol四氯化碳、12mmol催化剂1(r1和r2都为2-丁基)、220mmol苯依次加入反应瓶中,室温搅拌均匀,通氮气,于70℃下搅拌反应12h,停止加热,冷却至室温,向反应混合物中加入5ml水,搅拌后分出有机层,无水硫酸钠干燥,减压蒸去苯和未反应的四氯化碳,得式(iii)化合物,收率85%。该粗产品不用纯化,直接进行下一步反应。
[0037]
实施例4式(iii)化合物二苯基二氯甲烷的制备
[0038]
将100mmol四氯化碳、12mmol催化剂1(r1和r2都为2-氯原子)、220mmol苯依次加入反应瓶中,室温搅拌均匀,通氮气,于70℃下搅拌反应12h,停止加热,冷却至室温,向反应混合物中加入5ml水,搅拌后分出有机层,无水硫酸钠干燥,减压蒸去苯和未反应的四氯化碳,得式(iii)化合物,收率75%。该粗产品不用纯化,直接进行下一步反应。
[0039]
实施例5式(i)化合物4-甲氧苯基二苯基氯甲烷的制备
[0040]
将0.3g催化剂2、50ml二氯乙烷加入反应瓶中,室温搅拌0.5h,再加入50mmol苯甲醚和60mmol式(iii)化合物,于50℃下搅拌反应6h,停止加热,冷却至室温,过滤分离催化剂,催化剂晾干后可重复使用。向反应混合物中加入50ml水,搅拌后分出二氯乙烷层,水层再用二氯乙烷萃取2次,合并二氯乙烷层,无水硫酸钠干燥,减压蒸去溶剂,得粗固体,将粗固体用正己烷打浆,过滤固体,40℃减压干燥4h,得浅橙色式(i)化合物,收率88%,纯度为99%。
[0041]
实施例6式(i)化合物4-甲氧苯基二苯基氯甲烷的制备
[0042]
将0.4g催化剂2、50ml二氯乙烷加入反应瓶中,室温搅拌0.5h,再加入50mmol苯甲醚和60mmol式(iii)化合物,于60℃下搅拌反应6h,停止加热,冷却至室温,过滤分离催化剂,催化剂晾干后可重复使用。向反应混合物中加入50ml水,搅拌后分出二氯乙烷层,水层再用二氯乙烷萃取2次,合并二氯乙烷层,无水硫酸钠干燥,减压蒸去溶剂,得粗固体,将粗固体用正己烷打浆,过滤固体,40℃减压干燥4h,得浅橙色式(i)化合物,收率90%,纯度为
98.8%。
[0043]
实施例7式(i)化合物4-甲氧苯基二苯基氯甲烷的制备
[0044]
将0.15g实施例4中回收的催化剂2、30ml二氯乙烷加入反应瓶中,室温搅拌0.5h,再加入25mmol苯甲醚和30mmol式(iii)化合物,于50℃下搅拌反应6h,停止加热,冷却至室温,过滤分离催化剂,催化剂晾干后可重复使用。向反应混合物中加入30ml水,搅拌后分出二氯乙烷层,水层再用二氯乙烷萃取2次,合并二氯乙烷层,无水硫酸钠干燥,减压蒸去溶剂,得粗固体,将粗固体用正己烷打浆,过滤固体,40℃减压干燥4h,得浅橙色式(i)化合物,收率87%,纯度为99%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1