一种硝呋太尔的制备方法与流程
1.本发明涉及医药制造技术领域,具体涉及一种制备硝呋太尔的新方法。
背景技术:
2.硝呋太尔(nifuratel)是一种光谱抗菌素,可用于治疗由细菌、滴虫、霉菌和念珠菌引起的外阴、阴道感染和白带增多及泌尿系统感染,消化道阿米巴病及贾第虫病。其化学名为5-[(甲硫基)甲基]-3-[(5-硝基糠叉)氨基]-2-恶唑烷酮,cas登记号4936-47-4,结构式如下所示。
[0003][0004]
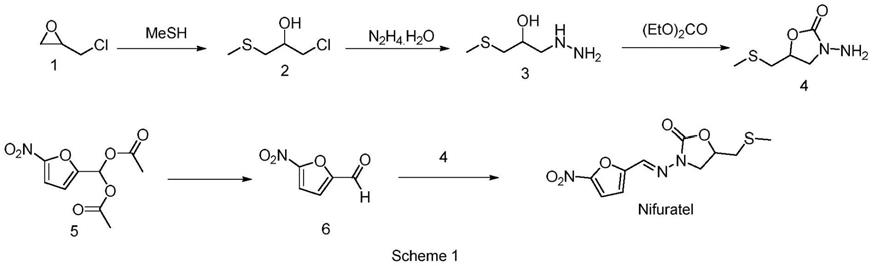
目前关于硝呋太尔的全合成有三条路线报道。第一条路线如scheme 1所示,以环氧氯丙烷为原料,其与甲硫醇反应得到中间体2;中间体2与水合肼反应制得中间体3,随后与碳酸二乙酯环合得到中间体4;化合物5经酸水解得到5-硝基糠醛,其与4反应制得硝呋太尔。该路线使用的甲硫醇,气味恶臭,污染环境;使用的水合肼要过量3倍以上,未参与反应的水合肼亦造成环境污染。
[0005][0006]
第二条路线如scheme 2所示,该路线与scheme 1所示方法相似,仅用甲硫醇钠代替了甲硫醇。该路线中合成的中间体7,沸点低,挥发性高,味道恶臭,污染环境,同时水合肼的过量使用也造成了环境污染。
[0007][0008]
第三条路线如scheme 3所示,以硫脲为反应起始原料,与硫酸二甲酯反应得到中
间体9;其与环氧氯丙烷反应得到中间体7;随后的路线与scheme 1和scheme 2相同。该合成方法亦产生了低沸点的化合物7,同时也使用了过量的水合肼以及剧毒的硫酸二甲酯。
[0009]
技术实现要素:
[0010]
为克服上述技术路线的不足,本发明提供了一种硝呋太尔合成的新方法,采用如下技术路线:
[0011][0012]
包括以下操作步骤:
[0013]
s1:化合物11的合成
[0014]
将化合物1溶于有机溶剂中,加入化合物10,随后在25℃下搅拌反应,tlc检测直至化合物10消失;向反应液中加入水,分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物11。
[0015]
其中,上述的有机溶剂为二氯甲烷、氯仿、四氢呋喃、1,4-二氧六环及甲基叔丁基醚中的一种或几种,优选四氢呋喃。
[0016]
其中,化合物1与化合物10的摩尔比为1:1~1.5,优选的,化合物1与化合物10的摩尔比为1:1~1.1。
[0017]
s2:化合物12的合成
[0018]
将化合物11溶于有机溶剂中,加入cdi,随后在一定温度范围内反应,tlc检测直至化合物11消失;向反应液中加入水,分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物12。
[0019]
其中,上述有机溶剂为四氢呋喃、乙腈、丙酮及甲苯中的一种或几种,优选四氢呋喃。
[0020]
其中,化合物11与cdi的摩尔比为1:1~2;优选的,化合物11与cdi的摩尔比为1:1~1.2。
[0021]
其中,反应温度为20~100℃,优选60~65℃。
[0022]
s3:化合物13的合成
[0023]
将化合物12溶于有机溶剂中,在0℃下滴加20%的甲硫醇钠水溶液;随后在25℃下搅拌反应,tlc检测直至化合物12消失;减压除去溶剂,加入二氯甲烷和水,分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,经甲基叔丁基醚重结晶得到化合物13。
[0024]
其中,上述有机溶剂为甲醇、乙醇、四氢呋喃、乙腈、二氯甲烷、氯仿及甲苯中的一种或几种,优选乙醇。
[0025]
其中,化合物12与甲硫醇钠的摩尔比为1:1~2,优选的,化合物13与甲硫醇钠的摩尔比为1:1~1.2。
[0026]
s4:化合物4的合成
[0027]
将化合物13溶于有机溶剂中,在0℃下加入酸;随后在25℃下搅拌反应,tlc检测直至化合物13消失;减压除去溶剂,向反应瓶中加入饱和碳酸氢钠水溶液,将体系ph调至7~8,加入二氯甲烷,分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物4;
[0028]
其中,上述有机溶剂为二氯甲烷、氯仿、四氢呋喃及甲苯中的一种或几种,优选二氯甲烷。
[0029]
其中,酸为盐酸或三氟乙酸,优选三氟乙酸。
[0030]
其中,三氟乙酸与有机溶剂的体积比为1:4~1:1,优选的,三氟乙酸与有机溶剂的体积比为1:4~1:2。
[0031]
s5:硝呋太尔的合成
[0032]
将化合物4溶于有机溶剂中,在0℃下加入5-硝基糠醛,随后在20℃下搅拌反应,tlc检测直至化合物4消失;抽滤,滤饼干燥,经乙醇重结晶后得硝呋太尔纯品。
[0033]
其中,有机溶剂为甲醇、乙醇、乙腈、四氢呋喃及二氯甲烷中的一种或几种,优选乙醇。
[0034]
其中,化合物4与5-硝基糠醛的摩尔比为1:1~1.5,优选的,化合物4与5-硝基糠醛的摩尔比为1:1~1.2
[0035]
与现有技术相比,本发明的有益效果包括:
[0036]
1、本发明提供了硝呋太尔合成的新方法,使用了新的合成原料肼基甲酸叔丁酯,避免过量水合肼的使用,同时未产生低沸点易挥发的含硫中间体,环境友好。
[0037]
2、该方法具有路线简短、原料廉价易得、产率和纯度高的优点,适合工业化生产。
具体实施方式
[0038]
为了使本发明的目的、技术方案及优点更加清楚明白,以下具体实施例对本发明进行进一步详细说明。本发明中的实验方法,如无特殊说明,均为常规方法。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0039]
本发明的反应进程可采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行监测,一般以原料消失时为反应终点。
[0040]
本发明的化合物1、化合物10、cdi及5-硝基糠醛购自上海毕得医药科技有限公司,其他化学试剂和原料均购自萨恩化学技术(上海)有限公司。
[0041]
实施例1:
[0042]
本发明的实施例1提供了一种中间体11的制备方法,其合成路线如下:
[0043][0044]
具体采用如下方法制备:
[0045]
将化合物1(120g,1.3mol)溶于四氢呋喃(1.5l)中,加入化合物10(171.4g,1.3mmol),随后在25℃下搅拌反应24h,tlc检测化合物1消失;向反应液中加入水(500ml),分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物11。
[0046]
采用本方法制备得到淡黄色油状物282g,收率96.8%。
[0047]
对本实施例中制得的中间体11进行鉴定,得到如下结果:
[0048]
esi-ms(m/z):225.6;
[0049]1h nmr(400mhz,cdcl3)δ7.06(d,j=5.7hz,1h),4.01-4.04(m,1h),3.54(dd,j=2.1,1.4hz,2h),3.46(d,j=4.2hz,1h),2.78(m,2h),1.39(s,9h).
[0050]
实施例2:
[0051]
本发明的实施例2提供了一种中间体12的制备方法,其合成路线如下:
[0052][0053]
具体采用如下方法制备:
[0054]
将化合物11(280g,1.25mol)溶于四氢呋喃中(2l)中,加入cdi(202g,1.25mol),随后在60℃下反应12h,tlc检测化合物11消失,向反应液中加入水(0.6l),分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物12。
[0055]
采用本方法得到的白色固体296g,收率94.8%。
[0056]
对本实施例中制得的中间体12进行鉴定,得到如下结果:
[0057]
esi-ms(m/z):251.7;
[0058]1h nmr(400mhz,cdcl3)δ7.76(s,1h),5.12
–
5.14(m,1h),3.79(d,j=0.9hz,2h),3.66(m,2h),1.42(s,9h).
[0059]
实施例3:
[0060]
本发明的实施例3提供了一种中间体12的制备方法:
[0061]
将化合物11(200g,0.9mol)溶于乙腈中(1.4l)中,加入cdi(144g,0.9mol),随后在60℃下反应12h,tlc检测化合物11消失,向反应液中加入水(0.4l)和乙酸乙酯(1l),分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物12。
[0062]
采用本方法得到的白色固体191g,收率85.6%。
[0063]
实施例4:
[0064]
本发明的实施例4提供了一种中间体13的制备方法,其合成路线如下:
[0065][0066]
具体采用如下方法制备:
[0067]
将化合物12(260g,1.04mol)溶于乙醇(1.5l)中,在0℃下滴加20%的甲硫醇钠溶液(363.5ml,1.04mol),随后在25℃下搅拌反应8h,tlc检测化合物12消失;减压除去乙醇,向体系中加入二氯甲烷(2l)和水(1.0l),分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,经甲基叔丁基醚(1.2l)重结晶得到化合物13。
[0068]
采用本方法得到的淡黄色固体259g,收率95.2%。
[0069]
对本实施例中制得的化合物13进行鉴定,得到如下结果:
[0070]
esi-ms(m/z):263.2;
[0071]1h nmr(400mhz,cdcl3)δ7.80(s,1h),5.05
–
5.09(m,1h),3.71(m,2h),3.06(m,2h),1.41(s,9h).
[0072]
实施例5:
[0073]
本发明的实施例5提供了一种中间体4的制备方法,其合成路线如下:
[0074][0075]
具体采用如下方法制备:
[0076]
将化合物13(240g,0.91mol)溶于二氯甲烷(0.85l)中,在0℃下加入三氟乙酸(272ml,3.66mol),随后在25℃下搅拌反应5h,tlc检测化合物13消失;减压除去溶剂,向反应瓶中加入饱和碳酸氢钠水溶液,将体系ph调至7~8,加入二氯甲烷(0.5l),分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物4。
[0077]
采用本方法得到的白色固体138g,收率93.0%。
[0078]
对本实施例中制得的化合物4进行鉴定,得到如下结果:
[0079]
esi-ms(m/z):163.3;
[0080]1h nmr(400mhz,cdcl3)δ4.65
–
4.70(m,1h),3.99(dd,j=12.5,2.4hz,1h),3.81(dd,j=12.4,2.3hz,1h),3.00
–
3.09(m,2h),2.12(s,3h).
[0081]
实施例6:
[0082]
本发明的实施例6提供了一种中间体4的制备方法:
[0083]
将化合物13(240g,0.91mol)溶于四氢呋喃(0.9l)中,在0℃下滴加盐酸溶液(4mol/l,0.92l,3.66mol),随后在25℃下搅拌反应2h,tlc检测化合物13消失;减压除去溶剂,向反应瓶中加入饱和碳酸氢钠水溶液,将体系ph调至7~8,加入二氯甲烷(0.5l),分液萃取,使用无水硫酸钠干燥有机层,抽滤,减压除去溶剂,即得化合物4。
[0084]
采用本方法得到的白色固体127g,收率85.6%。
[0085]
实施例7:
[0086]
本发明的实施例7提供了一种硝呋太尔的制备方法,其合成路线如下:
[0087][0088]
具体采用如下方法制备:
[0089]
将化合物4(130g,0.8mol)溶于乙醇(1.0l)中,在0℃下加入5-硝基糠醛(124g,0.88mol),随后在20℃下搅拌反应14h,tlc检测化合物4消失;抽滤,滤饼干燥,经乙醇重结晶后得硝呋太尔纯品。
[0090]
采用本方法得到黄色结晶状固体212g,收率92.7%,纯度99.8%。
[0091]
对本实施例中制得的硝呋太尔进行鉴定,得到如下结果:
[0092]
esi-ms(m/z):286.3;
[0093]1h nmr(400mhz,dmso-d6)δδ7.91(s,1h),7.76(d,j=4.9hz,1h),7.15(d,j=4.7hz,1h),4.80-5.10(m,1h),4.08
–
4.12(m,1h),3.82(dd,j=12.4,2.7hz,1h),2.80-3.05(m,2h),2.15(s,3h).
[0094]
实施例8:
[0095]
本发明的实施例8提供了一种硝呋太尔的制备方法:
[0096]
将化合物8(128g,0.79mol)溶于乙腈(0.8l)中,在0℃下加入5-硝基糠醛(122g,0.87mol),随后在20℃下搅拌反应10h,tlc检测化合物4消失;抽滤,滤饼干燥,经乙醇重结晶后得硝呋太尔纯品。采用本方法得到黄色结晶状固体196g,收率87.1%,纯度99.8%。
[0097]
以上所述本发明的具体实施方式,并不构成对本发明保护范围的限定。任何根据本发明的技术构思所做出的各种其他相应的改变与变形,均应包含在本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1