NIR/PET双模态造影剂及其制备方法与应用
本发明属于造影剂技术,具体涉及近红外和正电子发射计算机断层显像nir/pet双模态造影剂及其制备方法与应用。
背景技术:
由于早期诊断方法缺乏,肝癌患者被确诊时已为晚期,而晚期肝癌高度恶性且进展迅速,难以治疗,并且化疗药物的低选择性和低毒性导致治疗效果通常较差。因此,寻找有效的肝癌诊断方法,早发现,早治疗具有重要的临床应用价值。
作为非侵入性的检测方式,分子影像技术迅速发展,在肝癌诊断和治疗中发挥了非常重要的作用。目前,临床上使用的肝癌造影剂仅有钆塞酸二钠。钆塞酸二钠在正常肝细胞中摄取,而在肝癌细胞中不摄取,组织对比时在肝癌部位暗成像。然而钆塞酸二钠并非靶向肿瘤细胞,仅有的一个专一性造影剂不能满足目前临床对肝癌诊断的需求,能够靶向肝癌部位的造影剂是急需解决的难题。
多模态造影剂组合多种影像技术如pet,mri,nir等的优点,可以显著提高肿瘤组织成像的特异性和图像分辨率,为肝癌诊断实现靶向性和精准性提供可能。研究发现,近红外造影剂吲哚箐绿(indocyaninegreen,icg)进入血液后与血浆蛋白形成复合物,被动靶向至内皮网状细胞丰富的肝脏区域。随后,在有机阴离子转运多肽(organicaniontransportingpolypeptides,oatps)以及牛磺胆酸转运多肽(sodiumtaurocholatecotransportingpolypeptide,ntcp)介导下被细胞摄取。肝癌细胞膜上过度表达oatp与ntcp受体,且肝癌细胞胆管堵塞并缺乏有效的淋巴回流。最终,导致icg在正常肝细胞排出,而在肿瘤部位蓄积,可以实现肝癌部位的靶向性。患者在手术前注射icg,大约24小时后,肝脏表面的肿瘤借助近红外成像可以直观地呈现在外科医生面前,增加手术切除的准确性。但是,icg在肝癌检测中存在的最大挑战,就是组织穿透性差,只能检测位于10mm深度以上的肝癌,深部的肿瘤无法发现。相比于icg的近红外荧光成像,pet穿透力强能够检测组织内部的肿瘤,且灵敏度较高,但常规显像剂18f-fdg在肿瘤中聚集不够,且缺乏靶向肝癌细胞的能力。临床实践发现,探索pet/nir双模态探针分子策略,将肝癌靶向性、组织穿透力两方面的优势相结合,寻找敏感而高效的双模态造影剂候选化合物以期实现肝癌的精准定位,是早期发现肝癌患者,解决患者长期生存率低,满足临床诊疗需求的关键。
技术实现要素:
发明目的:针对上述现有技术中存在的技术问题,本申请提供了一种近红外和正电子发射计算机断层显像nir/pet双模态造影剂及其制备方法与应用。
技术方案:本申请所述的一种如式(i)所示的金属络合物:
其中,w1和w2相同或不同,各自独立地为放射性金属离子;
ligand-linker-nir-linker-ligand为配体;
条件是:当y1为-coo-或-so3-时,则halo-不存在。
其中,所述放射性金属离子选自64cu(2+)、68ga(3+)、90y(3+)、177lu(3+)、89zr(4+)、89sr(2+)、188re(2+)或225ac(3+)。
本申请中,ra1、ra2、ra3和ra4中,所述c1-4烷基优选为甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基。
本申请中,w1和w2优选相同,优选为68ga(3+)。
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
本申请中,
作为一种优选的技术方案,配体ligand-linker-nir-linker-ligand中,
进一步优选的,
本申请中,配体ligand-linker-nir-linker-ligand优选为如下任一化合物:
化合物通式1
具体如下:
备注:“/”表示无。
化合物通式2
具体如下:
备注:“/”表示无。
本申请中,所述的如式(i)所示的金属络合物优选为如下任一化合物:
化合物通式i-1
具体如下:
备注:“/”表示无。
化合物通式i-2
具体如下:
备注:“/”表示无。
本申请还提供了一种所述的如式(i)所示的金属络合物制备方法,其包括下列步骤:将配体与放射性金属淋洗液进行如下所示的反应,制得如式(i)所示的金属络合物:
其中,所述配体为ligand-linker-nir-linker-ligand;所述放射性金属淋洗液为放射性金属w1淋洗液和/或放射性金属w2淋洗液;所述的淋洗液中放射性金属的放射性活度为111~185mbq,所述的淋洗液中的溶剂为酸。
其中,各字母和基团的定义均同前所述。
所述的如式(i)所示的金属络合物制备方法中,所述配体优选以配体的醋酸钠缓冲液(优选0.25m)的形式参与到反应中。所述配体溶液优选在约100℃预热约5分钟后再使用。
所述的如式(i)所示的金属络合物制备方法中,所述酸优选无机酸,例如盐酸水溶液。所述酸的浓度为约0.05mol/l。
所述的如式(i)所示的金属络合物制备方法中,所述放射性金属淋洗液优选68ga淋洗液。
所述的如式(i)所示的金属络合物制备方法中,所述反应优选在约100℃下进行。
所述的如式(i)所示的金属络合物制备方法中,所述反应结束后,所得的反应液优选通过活化的sep-packc18纯化。其中洗脱液优选为60%的乙醇。
所述的如式(i)所示的金属络合物制备方法优选包括下列步骤:
(1)使用0.05mhcl对锗-镓发生器(itg公司)进行淋洗,淋洗流速1ml/min,按体积收集,1ml一管,将第2ml的68ga淋洗液作为反应溶液备用;
(2)取配体加入标记瓶中,以醋酸钠缓冲液(优选0.25m)溶解,100℃加热条件轻轻振荡混匀并预热5min,然后加入放射性活度为111~185mbq的68ga淋洗液,轻轻振荡混匀后100℃反应10min。
所述的如式(i)所示的金属络合物制备方法中,所述反应后溶液通过活化的sep-packc18小柱进行纯化,并经60%乙醇洗脱收集,以生理盐水稀释后保存备用,测定其放射性活度并计算标记产率,同时取少量置于37℃恒温箱内匀速振荡,4h后取20μl混合液,通过radio-hplc测定其体外稳定性。整个合成时间约25min,反应后所得产品的放射性活度除以反应前所加入68ga淋洗液的放射性活度,可得标记产率在90%以上。
所述的如式(i)所示的金属络合物制备方法,还可进一步包括配体ligand-linker-nir-linker-ligand的制备方法,其包括下列步骤:在碱和缩合剂的作用下,将如式(1-a)所示的化合物与ligand进行缩合反应制得配体ligand-linker-nir-linker-ligand,
linker-nir-linker+ligand→ligand-linker-nir-linker-ligand
(1-a)
其中,
ligand为
优选地,所述碱为无机碱和/或有机碱;所述的无机碱优选为碱金属碳酸盐或碱金属碳酸氢盐,例如为碳酸氢钠、碳酸氢钾、碳酸钠和碳酸钾中的一种或多种;所述的有机碱优选为三乙胺、二异丙基乙胺和吡啶中的一种或多种。条件是:当当所述碱仅为有机碱时,所述缩合反应结束后,所得的反应液需要在碱金属氢氧化物(例如氢氧化钠或氢氧化钾)的存在下,进行成盐反应,得到目标化合物。
优选地,所述缩合剂为环己基碳二亚胺、1-羟基苯并三唑、1-羟基-7-偶氮苯并三氮唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、2-(7-氧化苯并三氮唑)-n,n,n′,n′-四甲基脲六氟磷酸酯和o-苯并三氮唑-四甲基脲六氟磷酸盐中的一种或多种。
优选地,所述缩合反应在溶剂存在下进行,所述溶剂优选为酰胺类溶剂、酮类溶剂、腈类溶剂和亚砜类溶剂中的一种或多种;更优选地,所述溶剂为n,n-二甲基甲酰胺、丙酮、乙腈和二甲基亚砜中的一种或多种。
优选地,所述如式(1-a)所示的化合物与ligand的摩尔比为1:(1-10);优选为1:(2.3-2.5)。
优选地,所述如式(1-a)所示的化合物与碱的摩尔比为1:(1-10);优选为1:(3-3.5)。
优选地,所述如式(1-a)所示的化合物与缩合剂的摩尔比为1:(1-10);优选为1:(3-3.5)。
优选地,所述缩合反应的温度为-10-40℃,例如室温。
优选地,所述的制备方法中,反应的时间可不作具体限定,一般以tlc或hplc检测linker-nir-linker消失或不再进行反应时作为反应的终点。
具体制备过程如下:
将对肼基苯甲酸与3-甲基-2-丁酮经fisher吲哚合成反应得到化合物2。
化合物2与1,3-丙磺酸内酯或1,4-丁磺酸内酯反应得到3系列化合物。
3系列化合物与戊二烯醛缩二苯胺盐酸盐发生亲核加成消除反应得到5系列化合物。
三氯氧磷,n,n-二甲基甲酰胺,环己酮,苯胺反应得到4b。
3系列化合物与4b发生亲核加成消除反应得到6系列化合物。
linker系列化合物与二碳酸二叔丁酯反应生成单边boc保护化合物。
5系列化合物和6系列化合物分别与单boc保护的linker系列化合物反应生成7系列化合物和8系列化合物。
7系列化合物和8系列化合物通过三氟醋酸脱去boc得到9系列化合物和10系列化合物。
dota与溴乙酸叔丁酯反应生成do3a。
do3a与溴乙酸苄酯反应生成bn-do3a。
bn-do3a与钯碳和氢气反应生成do3a-cooh。
do3a-cooh和9系列化合物或10系列化合物经缩合得到t-npmc系列化合物。
t-npmc系列化合物脱去叔丁酯生成npmc系列化合物。
npmc系列化合物与68gacl3.6h2o反应得到终产物68ga-npc系列化合物
上述化合物中,l-表示
优选地,上述9系列化合物或10系列化合物的合成方法包括下列步骤:7系列化合物或8系列化合物在碱的存在下与linker系列化合物反应。所述碱优选为无机碱和/或有机碱。所述的无机碱优选碱金属碳酸盐或碱金属碳酸氢盐,例如为碳酸氢钠、碳酸氢钾、碳酸钠和碳酸钾中的一种或多种。所述的有机碱优选三乙胺、二异丙基乙胺和吡啶中的一种或多种。当所述碱仅为有机碱时,反应结束后,需要在碱金属氢氧化物的存在下,进行成盐反应,得到目标化合物。7系列化合物或8系列化合物与linker系列化合物的摩尔比优选为1:(1.3-1.5)。反应温度优选为室温。
优选地,t-npmc系列化合物的合成方法包括下列步骤:do3a-cooh化合物在碱和缩合剂的存在下与9系列化合物反应。终产物的合成方法中,溶剂优选为酰胺类溶剂、酮类溶剂、腈类溶剂和亚砜类溶剂中的一种或多种,更优选n,n-二甲基甲酰胺、丙酮、乙腈和二甲基亚砜中的一种或多种。所述碱优选为无机碱和/或有机碱。所述的无机碱优选碱金属碳酸盐或碱金属碳酸氢盐,例如为碳酸氢钠、碳酸氢钾、碳酸钠和碳酸钾中的一种或多种。所述的有机碱优选三乙胺、二异丙基乙胺和吡啶中的一种或多种。当所述碱仅为有机碱时,反应结束后,需要在碱金属氢氧化物的存在下,进行成盐反应,得到目标化合物。所述缩合剂优选为环己基碳二亚胺、1-羟基苯并三唑、1-羟基-7-偶氮苯并三氮唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯和o-苯并三氮唑-四甲基脲六氟磷酸盐中的一种或多种。9系列化合物与do3a-cooh化合物的摩尔比优选为1:(1-10),更优选为1:(2.3-2.5)。9系列化合物与碱的摩尔比优选为1:(1-10),更优选为1:(3-3.5)。9系列化合物与缩合剂的摩尔比优选为1:(1-10),更优选为1:(3-3.5)。9系列化合物与do3a-cooh化合物反应的温度优选为-10-40℃,例如室温。
优选地,终产物的合成方法包括下列步骤:10系列化合物在碱和缩合剂的存在下与do3a-cooh化合物反应。终产物的合成方法中,溶剂优选为酰胺类溶剂、酮类溶剂、腈类溶剂和亚砜类溶剂中的一种或多种,更优选n,n-二甲基甲酰胺、丙酮、乙腈和二甲基亚砜中的一种或多种。所述碱优选为无机碱和/或有机碱。所述的无机碱优选碱金属碳酸盐或碱金属碳酸氢盐,例如为碳酸氢钠、碳酸氢钾、碳酸钠和碳酸钾中的一种或多种。所述的有机碱优选三乙胺、二异丙基乙胺和吡啶中的一种或多种。当所述碱仅为有机碱时,反应结束后,需要在碱金属氢氧化物(例如氢氧化钠或氢氧化钾)的存在下,进行成盐反应,得到目标化合物。所述缩合剂优选为环己基碳二亚胺、1-羟基苯并三唑、1-羟基-7-偶氮苯并三氮唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯和o-苯并三氮唑-四甲基脲六氟磷酸盐中的一种或多种。10系列化合物与do3a-cooh化合物的摩尔比优选为1:(1-10),更优选为1:(2.3-2.5)。10系列化合物与碱的摩尔比优选为1:(1-10),更优选为1:(3-3.5)。10系列化合物与缩合剂的摩尔比优选为1:(1-10),更优选为1:(3-3.5)。10系列化合物与do3a-cooh系列化合物反应的温度优选为-10-40℃,例如室温。
优选地,终产物的合成方法优选包括下列步骤:将do3a-cooh化合物与缩合剂在冰浴下混合(活化),然后加入9系列化合物或10系列化合物的溶液,其中溶剂优选与反应溶剂相同,进行所述反应。
优选地,终产物的合成方法结束后,若碱选用的是有机碱,还包括后处理的操作。所述后处理的操作包括成盐与重结晶。所述成盐过程使用氢氧化钠与9系列化合物或10系列化合物的摩尔比优选为1:(1.05-1.15)。所述重结晶的溶剂为摩尔比为1:(15-25)的水与异丙醇混合溶液,甲醇与二氯甲烷混合溶液,甲醇与氯仿混合溶液或甲醇与乙酸乙酯混合溶液。
优选地,上述的68ga(3+)还可以替换成64cu(2+)、90y(3+)、177lu(3+)、89zr(4+)、89sr(2+)、188re(2+)、225ac(3+)等放射性金属离子。
优选地,上述的dota还可以替换成dtpa、nodaga、nota等。
本申请还提供了一种药物组合物,其包含所述如式(i)所示的金属络合物,以及药学上可接受的载体和/或赋形剂。
本申请还提供了一种所述如式(i)所示的金属络合物或所述的药物组合物在制备造影剂中的应用。所述的造影剂可用于诊断肿瘤如胃癌、肺癌、肝癌、食管癌、结肠直肠癌、恶性淋巴瘤、子宫颈癌、鼻咽癌或乳腺癌,优选用于诊断肝癌。所述的造影剂用于近红外(nir)成像和正电子发射型计算机断层显像(pet)成像中。因此,本申请的造影剂优选是一种nir和pet多模态造影剂。
本申请所述如式(i)所示的金属络合物,可配制静脉注射剂使用。
本申请中,pet是指正电子发射型计算机断层显像,通过标记上短寿命的放射性核素后,通过对于该物质在代谢中的聚集,来反映生命代谢活动的情况,从而达到诊断的目的。
本申请中,室温是指0-35℃。
有益效果:相比较于现有技术,本申请具有以下优势:(1)利用靶向择性富集于肿瘤(肝癌)区域,与周围正常组织形成较强对比的特点,与dota、dtpa等官能团相连接,在获取肿瘤(肝癌)靶向性的同时,增加穿透能力,设计合成了nir/pet双模态造影剂。体内外实验证实,本申请的造影剂水溶性好(水中溶解度可达到100mg/ml,可通过静脉注射给药)、毒性低,两种造影模态相互印证,丰富诊断信息,提高诊断精度,为临床上肝癌的早期诊断提供可行的新型造影剂。(2)本申请所制备的化合物可被肿瘤(肝癌)组织特异性吸收,使得该造影剂具有显著的肿瘤(肝癌)靶向性。(3)本申请所制备的化合物在nir/pet成像中的成像效果均与单模态阳性药效果类似,从而可结合两类造影手段的优势。
附图说明
图1是68ga-npc-3的radio-hplc图;
图2是68ga-npc-3在生理盐水中2h的radio-hplc图;
图3是68ga-npc-3在生理盐水中4h的radio-hplc图;
图4是荷载人源肝癌(hepg2)皮下肿瘤的裸鼠,尾静脉注射68ga-npc-3后15min进行micropet-ct融合显像图;
图5是荷载人源肝癌(hepg2)皮下肿瘤的裸鼠,尾静脉注射68ga-npc-3后进行micropet-ct融合显像图;
图6是荷载人源肝癌(hepg2)皮下肿瘤的裸鼠,尾静脉注射68ga-npc-3后120min进行micropet-ct融合显像图;
图7是荷载人源肝癌(lm3)原位癌的裸鼠,尾静脉注射68ga-npc-3后30min的micropet-ct显像和解剖后视觉观察图像。
具体实施方式
下面结合附图和实施例对本申请的内容作进一步地说明。
实施例1制备化合物68ga-npc-1,结构式如下:
反应路线如下:
步骤1:将4g4-肼基苯甲酸、3.96ml3-甲基-2-丁酮、4.32g醋酸钠、60ml醋酸投入250ml三颈瓶中,氮气保护;25℃搅拌3h,随后120℃反应6h;反应结束后,用水转移反应液,二氯甲烷(dcm)萃取,合并浓缩有机相;采用柱层析分离(dcm:甲醇=50:1),浓缩得到黄色固体化合物2,收率61%。
1hnmr(300mhz,cdcl3)δ(ppm):8.17(d,j=8.19hz,1h),8.08(s,1h),7.67(d,j=8.19hz,1h),2.41(s,3h,),1.40(s,6h)。
步骤2:向250ml三颈瓶中依次投入4g化合物2、11.93ml1,4-丁磺酸内酯、50ml邻二氯苯、氮气保护,180℃回流9h;反应结束后大量固体析出,抽滤,丙酮洗涤三次,得到粉红色固体化合物3,收率93%。
1hnmr(300mhz,dmso)δ(ppm):8.40(s,1h),8.17(dd,2h),4.52(t,2h),2.90(s,3h),2.51(t,2h),1.97(m,2h),1.77(m,2h),1.58(s,6h)。
步骤3:依次向250ml三颈瓶中加入2g化合物3、784mg戊二烯醛缩二苯胺盐酸盐、30ml醋酸酐、18ml冰醋酸,最后加入808.8mg醋酸钠,氮气保护,120℃回流45min;反应结束后,加入50ml无水乙醚,将析出的固体抽滤得到粗品,随后进行重结晶,溶剂选用摩尔比为4:1的异丙醇与水混合溶液,得到绿色化合物5a,收率76%。
1hnmr(300mhz,dmso)δ(ppm):8.08(d,2h,j=1.2hz),7.98(dd,2h,j=1.1,8.2hz),7.95(m,5h),7.51(d,2h,j=8.7hz),6.65(t,2h,j=12.4hz),6.54(d,2h,j=13.6hz),4.11(m,4h),3.09(m,4h),1.75(m,8h),1.67(m,12h)。
步骤4:在250ml三颈瓶中,将5.8ml乙二胺溶于15ml干燥的dcm中。冰浴,氮气保护,无水反应,开始搅拌。将3.2ml二碳酸二叔丁酯溶于65ml干燥的dcm中,缓慢滴加至反应体系。滴加完毕,撤除冰浴,换成25℃油浴反应18h。反应结束后,过滤除去副产物,向残留物中加入饱和碳酸氢钠溶液。用dcm萃取,合并浓缩有机相,得淡黄色油状物单boc-乙二胺,收率71%。
1hnmr(300mhz,cdcl3)δ(ppm):3.15(t,j=6.5hz,2h),2.77(t,j=6.5hz,2h),1.47(s,9h)。
步骤5:冰浴下,在250ml三颈瓶中,依次加入1.8g化合物5a、2.7g2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、80ml无水dmf、1.26mln,n-二异丙基乙胺。在冰浴下搅拌1h活化。1h后,加入单boc-乙二胺,加料完毕,撤出冰浴,在室温下搅拌12h。反应完毕后,用无水甲醇转移反应液,柱层析分离(dcm:甲醇=10:1;8:1;6:1;5:1;4:1)。浓缩至微量,加入大量dcm除杂,抽滤,得到绿色固体1.3g化合物7-1。收率52.8%。
1hnmr(300mhz,dmso-d6)δ(ppm):8.48(m,2h),8.09-7.84(m,7h,4ar-h,3–ch=ch),7.46(d,j=7.65hz,2h,2arh),6.94(m,2h),6.63-6.44(m,4h,4–ch=ch),4.09(br,4h,2n-ch2),3.11(m,8h),2.51(m,4h,2-ch2so3),1.86-1.76(m,8h,2n-ch2-ch2ch2-ch2-so3),1.66(12h,4-ch3),1.38(s,18h,9-ch3)。
步骤6:在50ml单颈瓶中,依次加入1g化合物7-1、3ml三氟醋酸、4ml无水dcm,氮气保护,室温下反应。反应结束后,用无水甲醇转移反应液,60℃下浓缩干燥,随后加入乙醚打浆。抽滤得到红色固体0.762mg化合物9-1。收率97.6%。
1hnmr(300mhz,dmso-d6)δ(ppm):8.53(m,2h),7.94-7.72(m,11h),7.47(m,2h,2arh),6.94(m,2h),6.63-6.44(m,4h,4–ch=ch),4.09(br,4h,2n-ch2),3.41(m,4h),2.88(m,4h),2.51(m,4h,2-ch2so3),1.86-1.76(m,8h,2n-ch2-ch2ch2-ch2-so3),1.64(s,12h,4-ch3)。
步骤7:在无水条件,冰浴下,在250ml三颈瓶中,将8.61g化合物dota、13.02g减压干燥的碳酸氢钠、100ml重蒸乙腈依次加入,最后缓慢滴加溴乙酸叔丁酯。反应完毕后,甲醇转移反应液,抽滤,将滤液在旋转蒸发仪上旋干,溶解于氯仿,用水萃取,合并有机相,用甲苯120℃重结晶,得到白色固体11.3g化合物do3a。收率44%。
1hnmr(300mhz,cdcl3)δ(ppm):10.14(br,s,1h,nh),3.39(br,s,4h),3.30(br,s2h,ch2),3.12(m,4h,ch2),3.09-2.89(m,12h,ch2),1.46(27h,9-ch3)。
步骤8:在无水条件下,在500ml三颈瓶中,将6.17g化合物do3a、4.98g干燥碳酸钾、300ml乙腈依次投入,氮气保护,室温下反应1h。1h后,将2.75ml溴乙酸苄酯缓慢滴加至反应体系内,30min内加完。反应24h。反应结束后,用dcm转移反应液,抽滤,将滤液旋干,得到黄色油状物。加入dcm溶解油状物,依次用水、饱和碳酸氢钠溶液、饱和食盐水洗涤,合并有机相,旋干的到红棕色油状物7g化合物bn-do3a。收率88%。
1hnmr(300mhz,cdcl3)δ(ppm):7.34-7.29(m,5h),5.12(s,2h),3.5-2.39(m,24h),1.46(27h,9-ch3)。
步骤9:氢气体系下,向500ml单颈瓶中依次加入8.2g化合物bn-do3a、1g钯碳、150ml乙醇,室温下反应12h,体系有气泡产生。反应结束后,在滤纸表面上铺上硅藻土防止漏碳,抽滤,将滤液浓缩得到油状物,加入dcm溶解,水洗2次,饱和碳酸氢钠洗两次,饱和食盐水洗两次,旋干溶剂,减压干燥得到4.5g白色固体化合物do3a-cooh。收率64.3%。
1hnmr(300mhz,cdcl3)δ(ppm):4.01-1.92(br,24h),1.48(27h,9-ch3)。
步骤10:冰浴条件下,向50ml三颈瓶中加入90mg化合物9-1、136mg化合物do3a-cooh、123.4mg2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、12mldmf。搅拌1h。1h后,加入90mg化合物9-1、0.08mln,n-二异丙基乙胺,撤除冰浴,室温下搅拌。反应完全后,旋干溶剂,加入乙醚析晶,抽滤得到固体,溶解制砂,柱层析分离(dcm:甲醇:千分之一tea=8:1-4:1),浓缩得72mg深绿色固体化合物。不进一步纯化,直接进行下一步反应。
步骤11:在250ml单颈瓶中,将步骤10所得粗品(1.64g,0.85mmol)溶于h2o:tfa=1:3(v/v)的混合溶液(60ml),加入三异丙基硅烷(0.7ml,3.4mmol),室温反应18h。tlc(dcm:甲醇=3:1)监测反应进程。反应完成后,旋除溶剂,乙醚逼出固体。制备型高效液相色谱分离,得到132mg蓝绿色粉末状固体,两步总收收率共计7.6%。
1hnmr(300mhz,dmso-d6)δ8.66(m,4h),8.15–7.82(m,7h),7.51(d,j=8.4hz,2h),4.16(br,4h),3.97(br,8h),3.80(br,8h),3.66(br,12h),3.45(br,12h),3.11(br,16h),2.53(m,4h),1.68(m,20h).
步骤12:制备化合物放射性68ga-npc-1,流程如下:
(1)使用0.05mhcl对锗-镓发生器(itg公司)进行淋洗,淋洗流速1ml/min,按体积收集,1ml一管,将第2ml的68ga淋洗液作为反应溶液备用;
(2)取50μg的标记前体npc-3加入标记瓶中,以1ml醋酸钠缓冲液(0.25m)溶解,100℃加热条件轻轻振荡混匀并预热5min,然后加入放射性活度为111~185mbq的68ga淋洗液,轻轻振荡混匀后100℃反应10min,反应后溶液通过活化的sep-packc18小柱进行纯化,并经60%乙醇洗脱收集,以生理盐水稀释后保存备用,测定其放射性活度并计算标记产率,同时取少量置于37℃恒温箱内匀速振荡,4h后取20μl混合液,通过radio-hplc测定其体外稳定性。
整个合成时间约25min,反应后所得产品的放射性活度除以反应前所加入68ga淋洗液的放射性活度,可得标记产率在90%以上。
68ga-npc-1的质控
hplc分析鉴定条件如下:高效液相色谱仪lc-20at(岛津公司)、色谱分析柱c18柱(4.6mm×250mm,zorbaxrax-c18柱)。流动相:a为含0.05%三氟乙酸(tfa)的水溶液;b为含0.05%tfa的乙腈溶液。梯度洗脱:梯度从5分钟的90%a和10%b增加到20分钟的20%a和80%b,流速为1ml/min。保留时间:14.9min,放射化学纯度:>99%。
实施例2制备化合物68ga-npc-3,结构式如下:
制备方法同实施例1,除了步骤4中用1,4-丁二胺代替乙二胺进行反应,其余合成步骤不变,得到终产品化合物68ga-npc-3,收率30%。
hrms(esi-tof)[(m+3h)3+]m/z:655.2003(calcdfor[m+3h]3+:655.2027relativeerror=0.37ppm).
实施例3制备化合物68ga-npc-4,结构式如下:
制备方法同实施例1,除了步骤4中用1,8-二氨基-3,6-二氧杂辛烷代替乙二胺进行反应,其余合成步骤不变,得到终产品化合物68ga-npc-4,收率30.2%。
hrms(esi-tof)[m+2nh4+na]3+m/z:713.9029(calcdfor[m+2nh4+na]3+:713.8959relativeerror=0.98ppm).
实施例4制备化合物68ga-npc-6,结构式如下:
反应路线:
制备的前两个步骤同实施例1,得到化合物3。
然后再制备nir信号分子结构,具体步骤如下:
步骤一:在250ml单颈瓶中,加入26mldmf,磁力搅拌,冰浴下滴加22mlpocl3。滴加完毕后,继续冰浴搅拌30min,撤除冰浴,加入11ml环己酮,氮气保护,加热回流1h。降至室温,改用机械搅拌,滴加36ml摩尔比1:1的苯胺与乙醇混合溶液。滴加完毕后,继续搅拌1h。加入220ml摩尔比10:1的为水和hcl的混合溶液,冰浴搅拌2h。抽滤,冰水、丙酮、乙醚洗涤滤饼。将固体打浆洗涤(pe:ea=2:1),得紫色固体4,收率32%。
1hnmr(300mhz,dmso)δ(ppm):8.40(s,2h),7.50-7.38(m,8h),7.21-7.16(m,2h),2.71(t,4h),1.80(m,2h)。
步骤二:在25ml单颈瓶中,依次加入173mg化合物3,87mg化合物4,68mg醋酸钠,1ml醋酸和2ml醋酸酐,氮气保护,120℃回流搅拌45min。溶液变为绿色,采用柱层析分离(pe:ea=2:1)监测至反应完全,停止加热,降至室温,将反应液倒入10ml乙醚中,绿色固体析出。抽滤,乙醚洗涤固体,柱层析分离(dcm:甲醇=3:1),得绿色固体6,收率46%。
1hnmr(300mhz,dmso)δ(ppm):8.27(d,j=14.2,2h),8.07(d,j=1.5,2h),7.97(d,j=1.6,2h),7.51(d,j=8.4,2h),6.60(d,j=13.8,2h),4.44–4.34(m,4h),2.58(d,j=6.7,4h),2.04(dt,4h),1.70(s,12h)。
得到绿色固体6后,继续按照实施例1中的方法,除了将化合物6代替化合物5a进行反应,其余合成步骤不变,得到终产品化合物68ga-npc-6,收率15.3%。
hrms(esi-tof)[m+2nh4+na]3+m/z:680.5066(calcdfor[m+2nh4+na]3+:680.5059relativeerror=0.98ppm).
实施例5制备化合物68ga-npc-14,结构式如下:
制备方法同实施例3,除了用1,3-丙磺酸内置代替1,4-丁磺酸内酯进行反应,其余合成步骤不变,得到终产品化合物68ga-npc-14,收率46.8%。
hrms(esi-tof)[(m+2h)+na]3+m/z:1039.2937(calcdfor[(m+2h)+na]3+:1039.2967relativeerror=0.29ppm).
实施例6制备化合物68ga-npc-16,结构式如下:
制备方法同实施例4,除了用1,3-丙磺酸内酯代替1,4-丁磺酸内酯与化合物2进行反应,其余合成步骤不变,得到终产品化合物68ga-npc-16,收率20.7%。
hrms(esi-tof)[(m+3h)3+]m/z:671.1633(calcdfor[m+3h]3+:671.5152relativeerror=0.20ppm).
实施例7制备化合物68ga-npc-19,结构式如下:
制备方法同实施例6,除了用1,8-二氨基-3,6-二氧杂辛烷代替乙二胺进行反应,其余合成步骤不变,得到终产品化合物68ga-npc-19,收率19.2%。
hrms(esi-tof)[(m+3h)3+]m/z:729.9966(calcdfor[m+3h]3+:729.5152relativeerror=0.20ppm).
nir/pet双模态造影剂的体内实验
本申请的细胞实验选用hepg2(人肝癌细胞)、l02(人正常肝细胞)与lm3(人肝癌细胞)细胞进行体外培养。所述的hepg2细胞购自中国科学院上海细胞所,l02细胞购自上海复生生物科技有限公司,所述的lm3细胞购自中国科学院上海细胞所。所述hepg2细胞在含有10%fbs,100iu/ml青霉素和100mg/ml链霉素的hyclonedmem培养基中培养。所述l02细胞在含有10%fbs,100iu/ml青霉素和100mg/ml链霉素的rpmi1640培养基中培养。所述lm3细胞在含有10%fbs,100iu/ml青霉素和100mg/ml链霉素的hyclonedmem培养基中培养。
本申请的动物实验中的模型鼠为普通裸鼠经腋下接种hepg2细胞,饲养裸鼠1周,得到肿瘤模型。
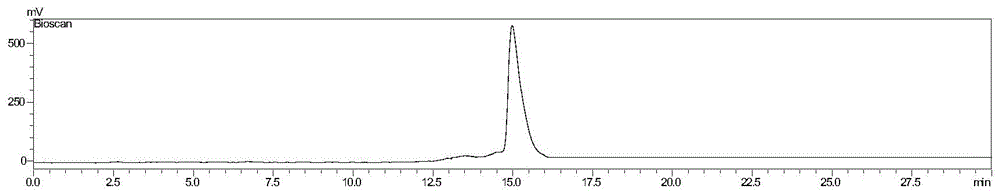
参见图1是68ga-npc-3的radio-hplc图,化合物保留时间为14.9分钟,化合物放射性化学纯度分别为(99.5%±0.2%)。
参见图2和图3是考察放68ga-nmc-3的体外稳定性的实验,化合物制备后置于生理盐水中,37℃孵育不同时间2h、4h。结果可见68ga-nmc-3在生理盐水中2h、4h内相对稳定,无明显脱标情况。
参见图4-图6是皮下荷载肝癌(hepg2)肿瘤的裸鼠,尾静脉注射68ga-npc-3后不同时间点micropet-ct显像。实验选取hepg2腋下接种裸鼠,经尾静脉注射放射性活度为3.7mbq的68ga-npc-3(0.1ml),在给药后不同时间(15min、60min、120min)对实验鼠进行micropet-ct融合成像,记录不同时间68ga-npc-3的药代动力学。图中圆圈部位指示肿瘤位置,肿瘤部位均可见明显放射性聚集,15min、60min、120min的肿瘤摄取(%id/g)分别为(4.2±0.2)、(1.8±0.05)、(0.7±0.06),肿瘤与本底(t/b)的比值分别为(3.5±0.2)、(3.7±0.3)、(5.1±0.7)。通过mciropet-ct动态显像,可见68ga-npc-3在荷瘤鼠体内不同脏器的的生物学分布,见附表1。
表1基于显像定量68ga-npc-3在荷瘤鼠体内不同脏器的的生物学分布(%id/g)
参见图7是荷载肝癌(lm3)原位癌的裸鼠,尾静脉注射68ga-npc-3后30min的micropet-ct显像和解剖后视觉观察图像,肝脏原位癌病灶对68ga-npc-3的摄取(%id/g为2.9±0.7),明显高于周围正常肝脏组织(%id/g为1.2±0.2)。显像剂主要经过肾脏和肝脏排泄,膀胱和肠道内可见68ga-npc-3的生理性排泄分布。
- 还没有人留言评论。精彩留言会获得点赞!