一种基于菲并咪唑的红光聚合物材料及其制备方法与流程
本发明属光电显示器件技术领域,具体涉及一种基于菲并咪唑的红光聚合物材料及其制备方法。
背景技术:
在电致发光领域,现有红光器件通常采用的重金属配合物会因自聚集而产生三线态-三线态的淬灭现象,导致发光效率很大的降低。因此提供一种器件效率高的红光电致发光材料成为了本领域技术人员亟待解决的技术问题。
技术实现要素:
本发明提供了一种基于菲并咪唑的红光聚合物材料及其制备方法,解决了现有技术缺少中红光器件发光效率低的技术问题。
本发明提供了一种基于菲并咪唑的红光聚合物材料,其结构式如(i)所示:
其中,r为氢、氘、卤素、c1-c20的直链或支链烷基,x=0-1,n=200-1000。
优选的,其结构式为:
优选的,x的取值范围为0.1-0.7。
本发明还提供了一种基于菲并咪唑的红光聚合物材料的制备方法,包括将式(ii)所示化合物,1-苯基-2-(4-(4,4,5,5-四甲基-1,3,2-二氧硼酯-2-基)苯基)-1h-菲并咪唑和式(v)所示化合物通过suzuki偶联反应制得式(i)所示聚合物;
优选的,所述suzuki偶联反应的温度为85℃。
优选的,式(ii)所示化合物通过以下步骤制得:
步骤1:将3,4-乙撑二氧噻吩通过溴化反应制得式(iv)所示化合物,
步骤2:将式(iv)所示化合物与4-碘苯酚通过取代反应生成式(iii)所示化合物,
步骤3:将2’,7’-二溴-9,9’-螺双[芴]-2,7-二(4-氨基苯基)酮、式(iii)所示化合物和1,10-菲罗啉通过取代反应制得式(ii)所示化合物。
本发明的有益效果如下:
本发明成功制备出红光聚合物,且其电致发光性能最佳数据为:启亮电压为2.2v,最大亮度为8697cdm-2,电流效率最高可达8.97cd/a,电致发光发射波长为644nm,色坐标为(0.62,0.37)。
附图说明
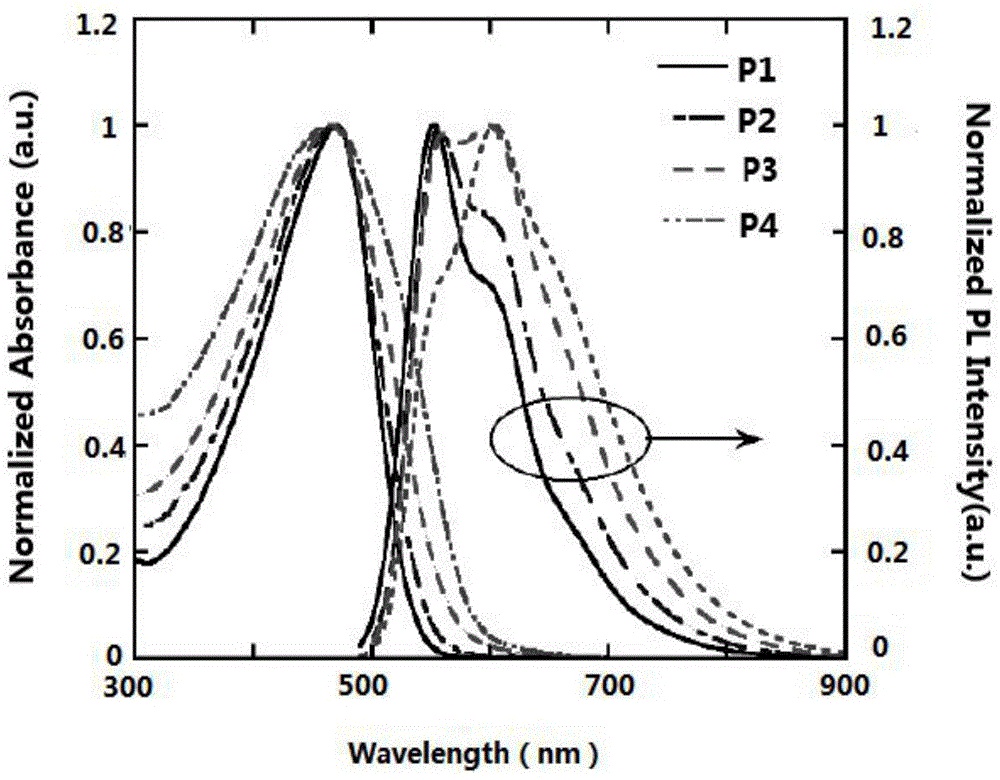
图1为本发明实施例制得聚合物的uv/pl发射光谱;
图2为本发明实施例制得的聚合物电致发光(el)光谱。
具体实施方式
下面结合具体实施例对本发明做进一步详细说明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1
在0-5℃下,向250ml的单口瓶内加入3,4-乙撑二氧噻吩(5ml,27.86mmol),冰醋酸(50ml)及浓硫酸(50ml),避光搅拌。然后分三次加(12.3g,180mmol)n-溴代丁二酰亚胺(nbs),再逐渐升至室温,反应过夜。用大量水稀释反应液,分离出固体,然后反复用nahco3水溶液和甲醇洗涤数次,晾干后用热氯苯溶剂纯化,制得式(iv)所示化合物(4.3g,产率70%),其化学反应方程式为:
实施例2
将150mldmf与式(iv)所示化合物(39.7g,180mmol),4-碘苯酚(25.36g,115.27mmol)和碳酸钾(31.74g,229.65mmol)中。将反应混合物在氮气下加热至80℃回流过夜。然后将混合物冷却至室温,并加入200ml冷水。随后将混合物用500ml二氯甲烷萃取。合并有机相,并用500ml饱和盐水洗涤,用硫酸镁干燥并蒸发溶剂。将取得的粗产物通过柱色谱法(石油醚)纯化,得到式(iii)所示化合物(46.6g,72%),其化学反应方程式为:
实施例3
将2,7-二溴-9,9'-螺双[芴](2.37g,5mmol)和氰化亚铜(3.13g,35mmol)置于50ml的两口瓶中,在氮气气氛下加热搅拌到完全溶解,向体系中加入1.5ml甲基磺酸,升温到140℃,混合溶液反应10小时。冷却后添加50ml无水乙醇,有白色固体析出,过滤,多次淋洗乙醇,干燥得到产物9,9'-螺双[芴]-2,7-二甲腈(1.55g,产率为85%),其化学反应方程式为:
实施例4
将9,9'-螺双[芴]-2,7-二甲腈(1.8g,30mmol)、铁屑(4.5g,80mmol)溶于100ml氯仿中,冰浴(0~5℃)下避光搅拌。小心取出液溴(15.5ml,301mmol),用100ml氯仿稀释后,将稀释后的液溴溶液缓慢加入到恒压滴液漏斗中,将恒压滴液漏斗中的液溴缓慢滴加到反应液中,室温下搅拌反应8h,产生的尾气用饱和亚硫酸氢钠水溶液处理。反应完毕,将反应液倒入冰的饱和亚硫酸氢钠水溶液中搅拌,然后过滤。滤渣用饱和亚硫酸氢钠洗涤处理,滤渣晾干后用乙醇重结晶纯化处理,过滤得到产物2',7'-二溴-9,9'-螺双[芴]-2,7-二碳腈(11.8g,产率75%),其化学反应方程式为:
实施例5
将2',7'-二溴-9,9'-螺双[芴]-2,7-二碳腈(5.24g,10mmol)和60ml氢氧化钠溶液加入100ml三口瓶中,在氮气气氛下加入12mg三(二亚苄基丙酮)二钯;将反应体系加热至100℃并反应10小时;反应完成后加入50ml水,搅拌20min后过滤,滤饼分别用甲醇(15ml)洗涤两次,随后烘干滤饼。将滤饼置于二氯甲烷(500ml)中,搅拌至完全溶解。随后减压浓缩溶液至15ml左右,析出大量固体,过滤固体,干燥滤饼后得到产物2',7'-二溴-9,9'-螺双[芴]-2,7-二羧酸(4.49g,产率为80%),其化学反应方程式为:
实施例6
将2mk2co3溶液(30ml)、2',7'-二溴-9,9’-螺双[芴]-2,7-二羧酸(5.62g,10mmol)和1-氯-4-硝基苯(5.9g,33mmol)加入到250ml三颈烧瓶中,加入100ml甲苯并引入氮气,温度升至70℃。停止反应,冷却至室温,浓缩反应溶液,用二氯甲烷萃取2-3次。将有机层旋转干燥后,用300-400目硅胶作为固定相,石油醚/二氯甲烷(1:4)作为洗脱剂,通过硅胶柱色谱纯化,最终得到产物2’,7’-二溴-9,9’-螺双[芴]-2,7-二(4-硝基苯基)酮(6.33g,产率82%),化学反应方程式如下:
实施例7
在氩气氛围下,往100ml三口烧瓶中加入2’,7’-二溴-9,9’-螺双[芴]-2,7-二(4-硝基苯基)酮(3.86g,5mmol)并用40ml甲苯溶解。再加入pd/c催化剂(98mg,0.11mmol)和30ml乙醇。在110℃下反应一天。冷却,倒入去离子水中。在混合液中加入适量的乙酸乙酯进行萃取,分液得到有机层,再用饱和的食盐水洗涤3~5次,然后有机层用无水硫酸镁干燥。干燥后通过抽滤得到滤液,在减压下蒸除溶剂。硅胶柱分离得产物2’,7’-二溴-9,9’-螺双[芴]-2,7-二(4-氨基苯基)酮(2.67g,产率75%),其化学反应方程式为:
实施例8
向2’,7’-二溴-9,9’-螺双[芴]-2,7-二(4-氨基苯基)酮(7.12g,10mmol)中加入式(iii)表示的化合物(10.8g,30mmol),氯化铜(0.05g,0.51mmol)和氢氧化钾(4g,71.3mmol)以及50ml甲苯。然后,将反应混合物冷却至室温,加入200ml水,用200ml二氯甲烷萃取3次,合并有机相并用水洗涤,用硫酸镁干燥,蒸发溶剂,粗产物用硅胶柱色谱纯化,使用石油醚:乙酸乙酯=10:1作为洗脱剂,得到式(ii)所示的化合物(7.17g,产率61%),化学反应方程式为:
实施例9
将9,10-菲醌(21g,100mmol),苯胺(36.6ml,200mmol),溴苯甲醛(18.5g,100mmol)和醋酸铵(38.5g,500mmol)加入到500ml圆底烧瓶中,再用300ml冰醋酸溶解。氮气保护下,110℃下搅拌回流4小时。将反应物倒入100ml冰水中,抽滤。将抽滤所得固体以石油醚:二氯甲烷=1:1为洗脱剂经柱层析分离,乙醇重结晶,真空烘箱烘干,得到2-(4-溴苯基)-1-苯基-1h-菲并咪唑(34g,产率75%),其化学反应方程式为:
实施例10
将2-(4-溴苯基)-1-苯基-1h-菲并咪唑(3.73g,10mmol),联硼酸频哪醇酯(7.62g,30mmol),醋酸钾(2.94g,30mmol),[1,1’-双(二苯基膦基)二茂铁]二氯化钯(0.2g,1mmol)加入到500ml圆底烧瓶中,并溶解于50ml二氧六环中,在氮气保护,90℃下搅拌回流72小时。反应结束后,用二氯甲烧萃取,以石油醚:二氯甲烷=1:2为洗脱剂通过硅胶柱层析分离,旋转蒸发浓缩萃取液,真空烘箱烘干,得到1-苯基-2-(4-(4,4,5,5-四甲基-1,3,2-二氧硼酯-2-基)苯基)-1h-菲并咪唑(3.48g,产率62%),其化学反应方程式为:
实施例11
将1-苯基-2-(4-(4,4,5,5-四甲基-1,3,2-二氧硼酯-2-基)苯基)-1h-菲并咪唑(0.84g,2mmol),式(v)所示化合物(0.57g,0.90mmol和式(ii)所示化合物(0.116g,0.1mmol)加入到40ml三氯甲烷中,氩气气氛中加热搅拌,15min后加入12mg醋酸钯,温度稳定在85℃时加入1ml浓度为20%的四乙基羟氨水溶液,30min后停止通气,氩气气氛中反应36h。之后加入溴苯(125mg,0.8mmol)封端并继续反应4h。将反应停止并冷却到室温,沉析到250ml甲醇中,过滤干燥后索氏提取(先后用甲醇,丙酮和正己烷各抽提12h)。之后将聚合物干燥,用甲苯溶解,以200-300目硅胶为固定相,石油醚:二氯甲烷=4:1为洗脱剂进行柱层析,将溶液浓缩后再次沉析到甲醇中,固体过滤后放入真空干燥箱干燥,得到聚合物p1(2.36g,产率75%),其化学反应方程式为:
实施例12
本实施例12与实施例11的区别为:式(ii)所示化合物的含量为(0.36g,0.3mmol),式(v)所示化合物的含量为(0.44g,0.70mmol),制得聚合物p2(4.98g,产率74%),其化学式为:
实施例13
本实施例13与实施例11的区别为:式(ii)所示化合物的含量为(0.60g,0.5mmol),式(v)所示化合物的含量为(0.31g,0.5mmol),最终制得聚合物p3(6.89g,产率72%),其化学式为:
实施例14
本实施例14与实施例11的区别为:式(ii)所示化合物的含量为(0.84g,0.7mmol),式(v)所示化合物的含量为(0.186g,0.3mmol),最终制得聚合物p4(8.89g,产率71%),其化学式为:
综上,图1为本发明实施例制得聚合物uv/pl发射光谱,其中,紫外-可见吸收光谱(uv-vis)通过日本shimadzuuv-2600分光光度计测定,荧光发射光谱(pl)通过美国perkin-elmerls45x和日立f-4600荧光分光光度计测定。图中在410-420m左右的吸收峰为螺芴以及菲并咪唑主链的π-π*跃迁产生的。随着式(ii)所示化合物含量的增加,聚合物的pl光谱的波长从600nm移至620nm,均位于红光波长区域,其中表1为本发明实施例的光物理数据。
表1本发明实施例的光物理数据
由表1可知,本发明实施例均位于红光区域,具有较长的荧光寿命,且荧光量子产率较高,均超过85%。
本发明以ito/pedot:pss/polymer/csf/al为单层器件,在电流密度为12ma/cm2下测试聚合物的电致发光(el)光谱,其中,el光谱通过orielinstaspecivccdspectrograph测定,电流密度-电压采用keithley2420和konicaminoltachromametercs-200测试;eqe依据亮度、电流密度和电致光谱进行测定。如图2所示,聚合物中式(ii)所示化合物含量越高,电致发光光谱逐渐红移,这是由于聚合物中含有吸电子基团羰基,引入到聚合物主链后对lumo能级降低,使聚合物的带隙变小,均为红光聚合物,其中,表2为本发明实施例的电致发光数据。
表2本发明实施例的电致发光性能数据
由表2可知,本发明实施例14制备的器件其电致发光性能最佳,启亮电压最低为2.2v,最大亮度为8697cdm-2,电流效率最高可达8.97cd/a,电致发光发射波长为644nm,色坐标为(0.62,0.37)。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!