一种丁醚脲杂质D的合成方法与流程
本发明涉及药物化学领域,尤其涉及一种丁醚脲杂质d的合成方法。
背景技术:
丁醚脲(又名杀螨隆,结构如下化合物i)是汽巴-嘉基公司80年代开发出的一种新型硫脲杀虫、杀螨剂。其具有触杀、胃毒、内吸和熏蒸作用,且具有一定的杀卵效果。在紫外光下转变为具有杀虫活性的物质,对蔬菜上已产生严重抗药性的害虫具有较强的活性。可防治多种作物和观赏植物上的蚜虫、粉虱、叶蝉、夜蛾科害虫及害螨。
丁醚脲杂质d(结构如下化合物a)是生产丁醚脲过程中所产生的副产物,由于其含量较低,用提纯的方式很难将其富集。目前尚没有文献报道其合成方法。这给生产和申报农药工作带来了很多不便。
为了规范地进行对杂质进行研究,并将其控制在一个安全、合理的限度范围之内,提高丁醚脲的质量及安全性,提供丁醚脲杂质d的合成方法具有重要意义。
技术实现要素:
本发明为提供丁醚脲杂质d用于丁醚脲生产中杂质的定性及定量分析,从而可以提高丁醚脲的质量标准,为人民群众安全用药提供重要的指导意义,提供了丁醚脲杂质d的制备方法。
本发明提供了丁醚脲杂质d的制备方法,包括以下步骤:
步骤s1:将2,6-异丙基苯胺溶于dmf中,冷却后滴加n-溴代琥珀酰亚胺(nbs)的n,n-二甲基甲酰胺得到第一反应液,低温下反应完全,与水和醋酸乙酯混合后分液,得到第一有机相,所述第一有机相经多次水洗、干燥、旋干得化合物2;
步骤s2:将所述化合物2、碳酸钾、苯酚、氯化亚铜、1-甲基咪唑、二甲苯依次混合得到第二反应液,加热回流至反应完全,冷却后加入水和甲基叔丁基醚搅拌,分液得第二有机相,所述第二有机相经碱洗、水洗、干燥、旋干、柱层析得化合物3;
步骤s3:将所述化合物3、三乙胺、h2o依次混合后,缓慢滴加cs2得到第三反应液,第一温度搅拌反应一段时间后,升温至第二温度至反应完全,冷却后经ch2cl2多次萃取得到第三有机相,所述第三有机相经干燥、旋蒸、柱层析得到化合物a,即丁醚脲杂质d。
优选地,步骤s1中,所述低温为0~5℃。
优选地,步骤s2中,所述化合物2与所述苯酚的摩尔比为1:1~5。
优选地,步骤s2中,所述化合物2、所述碳酸钾、所述氯化亚铜的摩尔比为1:2~2.5:0.05~0.1。
优选地,步骤s2中,所述化合物2、1-甲基咪唑、二甲苯的质量体积比为1:0.1~0.2:8~15。
优选地,步骤s2中,所述碱洗采用5%氢氧化钠水溶液。
优选地,步骤s3中,所述化合物3与所述cs2的摩尔比为:1:0.3~0.6。
优选地,步骤s3中,所述化合物3与所述三乙胺的摩尔比为:1:1~1.5。
优选地,步骤s3中,所述化合物3与所述h2o的质量体积比为1:20~25。
优选地,步骤s3中,所述第一温度为室温;所述第二温度为100~120℃。
本发明采用以上技术方案,与现有技术相比,具有如下技术效果:
本发明为提供了丁醚脲杂质d的制备方法,以2,6-异丙基苯胺、苯酚、cs2等为原料,为规范地进行对杂质进行研究提供了物质基础,还可以用于丁醚脲生产中杂质的定性及定量分析,并将其控制在一个安全、合理的限度范围之内,从而可以提高丁醚脲的质量标准,为人民群众安全用药提供重要的指导意义。
附图说明
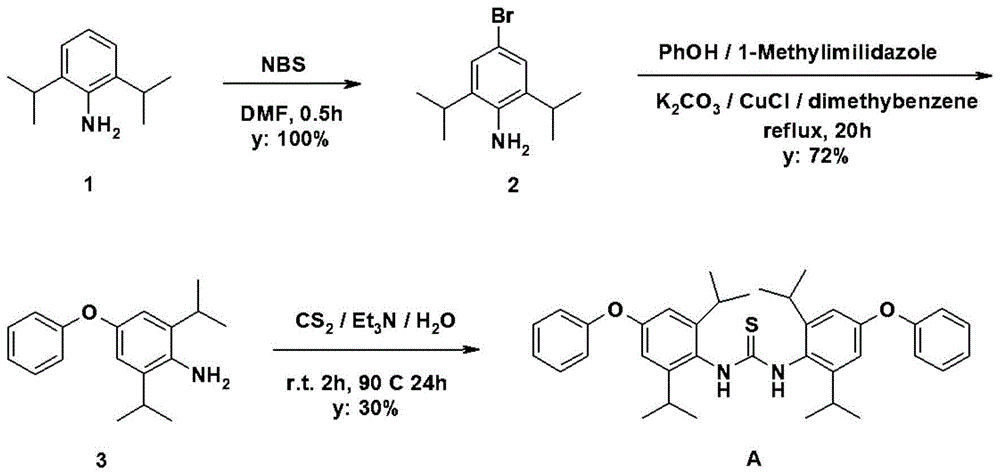
图1为本发明一种丁醚脲杂质d的制备方法的反应式;
图2为对丁醚脲杂质d进行核磁氢谱检测得到的谱图;
图3为对丁醚脲杂质d进行电喷雾质谱检测得到的谱图。
具体实施方式
本发明提供了丁醚脲杂质d的制备方法,如图1中,包括以下步骤:
步骤s1:将2,6-异丙基苯胺溶于dmf中,冷却后滴加n-溴代琥珀酰亚胺(nbs)的n,n-二甲基甲酰胺得到第一反应液,低温下反应完全,与水和醋酸乙酯混合后分液,得到第一有机相,所述第一有机相经多次水洗、干燥、旋干得化合物2;
步骤s2:将所述化合物2、碳酸钾、苯酚、氯化亚铜、1-甲基咪唑、二甲苯依次混合得到第二反应液,加热回流至反应完全,冷却后加入水和甲基叔丁基醚搅拌,分液得第二有机相,所述第二有机相经碱洗、水洗、干燥、旋干、柱层析得化合物3;
步骤s3:将所述化合物3、三乙胺、h2o依次混合后,缓慢滴加cs2得到第三反应液,第一温度搅拌反应一段时间后,升温至第二温度至反应完全,冷却后经ch2cl2多次萃取得到第三有机相,所述第三有机相经干燥、旋蒸、柱层析得到化合物a,即丁醚脲杂质d。
下面通过具体实施例对本发明进行详细和具体的介绍,以使更好的理解本发明,但是下述实施例并不限制本发明范围。
实施例1
本实施例提供了化合物2的制备方法(即步骤s1):
取2,6-异丙基苯胺(化合物1,27g,0.15mol),溶于n,n-二甲基甲酰胺(dmf,250ml)中,降温至0-5℃得到第一反应液,滴加n-溴代琥珀酰亚胺(nbs31.5g,0.18mol)的dmf(200ml)溶液,滴加完成后,0-5℃下反应0.5小时,tlc检测确认反应完全,加入水和乙酸乙酯搅拌10分钟,分液,有机相水洗两次,干燥,浓缩,得到化合物2(36g,收率100%);
对制备的化合物2进行核磁氢谱检测:
1hnmr(300mhz,dmso)δ1.10(d,j=6.9hz,12h),2.93-3.04(m,2h),4.76(s,2h),6.93(s,2h)
实施例2
本实施例提供了化合物3的制备方法(即步骤s2):
将化合物2(3g,0.011mol)、碳酸钾(3.24g,0.023mol)、苯酚(1.32g,0.014mol)、氯化亚铜(0.058g,0.581mmol)、0.47ml1-甲基咪唑、30ml二甲苯依次加入到三口瓶中得到第二反应液,加热回流反应12h,冷却后加入水和甲基叔丁基醚(mtbe)搅拌10min,分层得第二有机相,第二有机相用5%氢氧化钠水溶液洗一次、水洗一次、用无水硫酸钠干燥、旋干、柱层析得化合物3(2.3g,收率:72%)。
对制备的化合物3进行核磁氢谱检测:
1hnmr(300mhz,dmso)δ1.11(d,j=6.9hz,12h),2.98-3.09(m,2h),4.50(s,2h),6.60(s,2h),6.79-6.86(m,2h),6.93-7.01(m,1h),7.23-7.32(m,2h)
实施例3
化合物3(2.69g,10mmol),三乙胺(1.24g,12mmol),60mlh2o依次加入到100ml三口瓶中,然后缓慢滴加cs2(410mg,5.4mmol)得到第三反应液,室温搅拌反应2h后升温至100℃反应24h,反应结束后,冷却,ch2cl2萃取三次,得到第三有机相,第三有机相用无水硫酸钠干燥,旋蒸,柱层析纯化得产品化合物a,即丁醚脲杂质d(3.43g,收率:30%)。
对制备的化合物a进行核磁氢谱和电喷雾质谱检测(如图2-3中所示):
1hnmr(300mhz,cdcl3)δ1.01-1.23(m,24h),3.01(m,2h),3.32(m,2h),6.46(s,1h),6.72(s,2h),6.84(s,2h),6.9-7.32(m,10h),8.5(s,1h)
esi-ms:581(m+1),579(m-1)
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!