一种培比洛芬的制备工艺
本发明属于化学制药领域,具体涉及一种培比洛芬的制备工艺。
背景技术:
培比洛芬(pelubiprofen)属于苯丙酸类非甾体抗炎药,其化学名称为:(+)-(e)-2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸,结构式如下:
培比洛芬的合成方法目前有以下几种途径:
美国专利us4673761公开了由1-吗啉环己烯与2-(对甲酰基苯基)丙酸缩合反应,然后水解得到产物,最后通过乙酸乙酯和正己烷重结晶,收率81%。
美国专利us4365076公开了环庚酮与2-(4-甲酰基苯基)丙酸乙酯反应,然后水解,柱层析得到培比洛芬。该方法使用强碱,例如氢氧化钠、甲醇钠或叔丁醇钾等,从而不可避免的导致双缩合和二聚等副反应的发生,同时2-(4-甲酰基苯基)丙酸乙酯结构中存在对强碱敏感的酯基,导致反应的收率极低,不到40%,因此也没有工业上应用的价值。
近年来,直接构建碳-碳双键法被认为是理想的方法,因为它不需要活化的反应物做原料,另外不需要多步转化和从多种副产物进行分离而显著符合节能和原子经济性的要求。因此用未经活化的环己酮与2-(4-甲酰基苯基)丙酸酯反应直接高效选择性的构建碳-碳双键就变得非常具有吸引力,但是这种直接构建方法需要理想合适的催化剂。在合成催化领域,双协同有机催化剂得到了越来越多的重视,该类催化剂包含软的路易斯酸和软路易斯碱,可用于醛酮直接缩合得到а,-不饱和醛酮,并且能够避免竞争性的副产物,且对碱敏感基团有良好的兼容性。基于此,本发明通过在有机催化剂存在下直接构建碳-碳双键,高效的对培比洛芬关键合成步骤优化,从而开发一种新的培比洛芬合成工艺。具体的合成步骤如下:
技术实现要素:
本发明的目地在于提供一种培比洛芬的制备工艺,该工艺具有起始原料易得,操作简单、工艺稳定、收率高、产品质量好的特点。
为实现上述目的,本发明通过以下技术方案实现:
一种培比洛芬的制备工艺,包括以下步骤:
步骤(1):2-(4-溴甲基苯基)丙酸在有机溶液中,与甲醇反应得到2-(4-溴甲基苯基)丙酸甲酯(2);
步骤(2):步骤(1)中的产物在有机溶液中,在lewis酸的存在下,和六亚甲基四胺反应,得到2-(4-甲酰基苯基)丙酸甲酯(3);
步骤(3):步骤(2)中的产物在双协同有机催化剂存在下,和环己酮反应,得到缩合产物2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯(4)。
步骤(4):步骤(3)中的产物经水解反应,得到产物培比洛芬(5)。
所述步骤(1)中2-(4-溴甲基苯基)丙酸和甲醇的体积比为1∶1~5,最优选择为1∶3;反应温度为0℃~60℃,最优选择10℃。反应时间为1~24h,反应催化剂可以是浓硫酸或固体超强酸。优先采用固体超强酸。
在步骤(2)中2-(4-溴甲基苯基)丙酸甲酯和六亚甲基四胺的摩尔比为1∶1~5,最优选择1∶2,反应温度为20℃~100℃,最优选择80℃,反应时间为3~10h,反应溶剂可以为乙醇、甲醇、水、乙酸乙酯、四氢呋喃或上述溶剂任选所组成的混合溶剂。优先采用乙醇和水混合溶液。
在步骤(3)中化合物(3)和环己酮的摩尔比为1:(1~10);化合物3与催化剂之间的摩尔比为(5~10):1,催化剂和共催化剂的摩尔比为1:(1~3),反应温度为20~80℃,最优选择50℃~60℃,反应时间为6~48h,最优选择24h,反应催化剂可以是n,n-二甲基乙二胺、n,n-二乙基乙二胺、l-脯氨酸、5,5-二甲基-l-脯氨酸、四氢吡咯,优选催化剂为n,n-二甲基乙二胺;共催化剂可以是1,3-二甲基巴比妥酸和丙二酸亚异丙酯;反应溶剂可以是乙腈、二氯甲烷、氯仿、四氢呋喃、乙酸乙酯、甲醇或上述溶剂任选所组成的混合溶剂。优先采用乙酸乙酯。
在步骤(4)中化合物4在碱性条件下在醇中10~30℃反应,反应时间为10~24h,得化合物5;反应所用的碱可以是氢氧化钠溶液、氢氢化钾溶液、碳酸钾溶液或碳酸钠溶液,优选10%的碳酸钠溶液。
本发明以来源易得的2-(4-溴甲基苯基)丙酸为起始原料,第一步为经典的酯化反应,反应条件温和,操作简便,收率高;第二步为2-(4-溴甲基苯基)丙酸甲酯的醛基化,所用的反应原料六亚甲基四胺可在酸性条件下反应,后处理容易;第三步反应为双协同有机催化剂存在下的缩合反应,反应副反应少,后处理简单;第四步水解反应条件温和,反应收率较高。本发明提供了一种新的培比洛芬的合成工艺,适合工业化。
附图说明
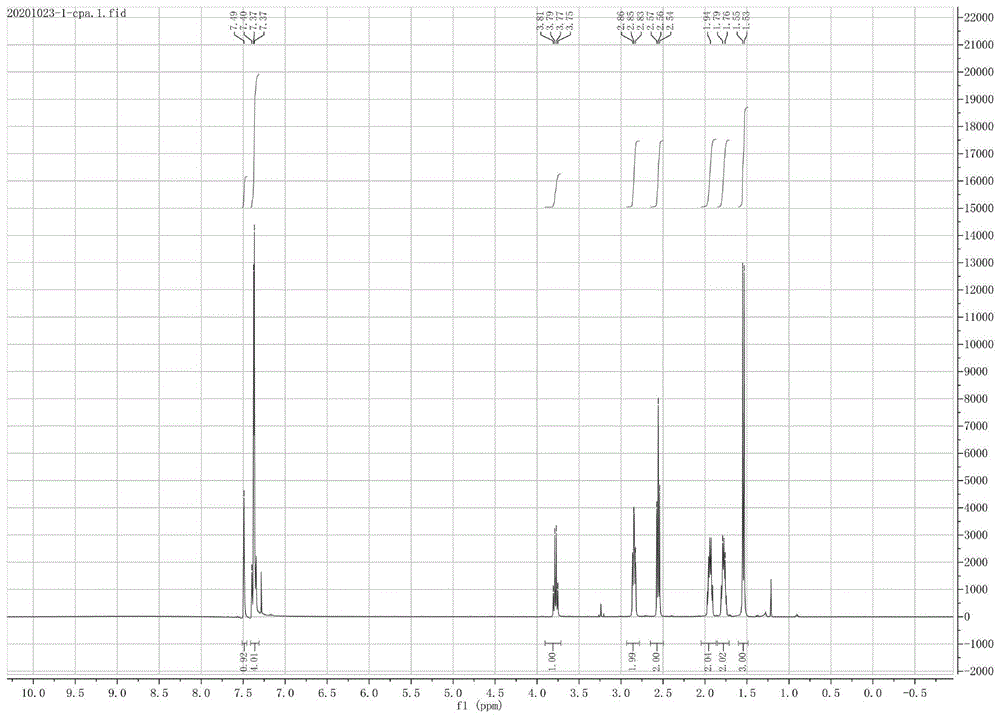
图1为核磁氢谱;图2为核磁碳谱。
具体实施方式
下面通过具体实施方式的描述对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以做出各种修改或者改进,但是只要不脱离本发明的基本思想,均在本发明的范围之内。
本发明实施例中所用的各种原料和试剂如无特别说明均为市售购买。
实施例1:一种培比洛芬的制备工艺,包括以下几个步骤:
步骤1:制备2-(4-溴甲基苯基)丙酸甲酯
取2-(4-溴甲基苯基)丙酸(32g)溶于甲苯(100ml)溶液中,依次加入甲醇(38g),浓硫酸(17g),控温在30℃反应5小时,反应毕,冷却分层,有机相经碱洗、水洗、干燥后减压浓缩,得33.4g化合物2-(4-溴甲基苯基)丙酸甲酯,橙黄色透明油状液体,纯度98%以上。
步骤2:制备2-(4-甲酰基苯基)丙酸甲酯
在反应瓶中加入24.5g六亚甲基四胺和水50ml,搅溶,加入30g2-(4-溴甲基苯基)丙酸甲酯和220ml乙醇,升温回流反应5小时;滴加45ml精制盐酸,继续反应45分钟。冷却,减压浓缩至干,加入100ml水和300ml甲苯,搅拌10分钟,静置分层,有机相经水洗、干燥后减压浓缩,得20g2-(4-甲酰基苯基)丙酸甲酯,黄色油状物,纯度97%以上。
步骤3:制备2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯
取2-(4-甲酰基苯基)丙酸甲酯(20mmol)溶于乙酸乙酯(20ml)溶液中,依次加入n,n-二甲基乙二胺(0.2mmol)和丙二酸亚异丙酯(0.4mmol),50℃下搅拌反应12h,降至室温,经水洗、干燥后减压浓缩,得淡黄色油状物2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯,收率81%,纯度95%以上。
步骤4:制备培比洛芬
向反应瓶中加入27.2g化合物2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯和200ml甲醇,搅拌溶解,室温下滴加103.5g碳酸钾溶液;滴毕,搅拌反应12小时。反应毕,用110g稀盐酸调节ph=3~5;用300ml二氯甲烷分3次萃取,合并有机相,经水洗、干燥,减压浓缩,得25g培比洛芬,黄色油状物,纯度90%以上。加入60g混合溶剂,重复精制2~3次,得培比洛芬成品15g,类白色结晶粉末,m.p106~110℃,纯度99%以上(核磁氢谱见附图1,核磁碳谱见附图2)。
1hnmr(400mhz,chloroform-d)δ7.49(s,1h),7.45–7.32(m,4h),3.78(q,j=7.2hz,1h),2.92–2.79(m,2h),2.61–2.50(m,2h),2.01–1.88(m,2h),1.85–1.72(m,2h),1.54(d,j=7.2hz,3h).13cnmr(101mhz,cdcl3)δ202.10,179.80,140.26,136.67,135.30,134.73,130.67,127.64,45.17,40.27,28.94,23.83,23.31,18.03.
实施例2:一种培比洛芬的制备工艺,包括以下几个步骤:
步骤1:制备2-(4-溴甲基苯基)丙酸甲酯
取2-(4-溴甲基苯基)丙酸(32g)溶于甲苯(100ml)溶液中,依次加入甲醇(38g),浓硫酸(17g),控温在20℃反应8小时,反应毕,冷却分层,有机相经碱洗、水洗、干燥后减压浓缩,得30.3g化合物2-(4-溴甲基苯基)丙酸甲酯,橙黄色透明油状液体,纯度98%以上。
步骤2:制备2-(4-甲酰基苯基)丙酸甲酯
在反应瓶中加入24.5g六亚甲基四胺和水50ml,搅溶,加入30g2-(4-溴甲基苯基)丙酸甲酯和200ml乙醇,升温回流反应3小时;滴加45ml精制盐酸,继续反应60分钟。冷却,减压浓缩至干,加入100ml水和300ml甲苯,搅拌20分钟,静置分层,有机相经水洗、干燥后减压浓缩,得21g2-(4-甲酰基苯基)丙酸甲酯,黄色油状物,纯度97%以上。
步骤3:制备2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯
取2-(4-甲酰基苯基)丙酸甲酯(20mmol)溶于乙酸乙酯(30ml)溶液中,依次加入n,n-二甲基乙二胺(0.25mmol)和二甲基巴比妥酸(0.5mmol),50℃下搅拌反应12h,降至室温,经水洗、干燥后减压浓缩,得淡黄色油状物2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯,收率82%,纯度95%以上。
步骤4:制备培比洛芬
向反应瓶中加入27.2g化合物2-[4-(2-(氧代-环己叉基甲基)-苯基]丙酸甲酯和150ml甲醇,搅拌溶解,室温下滴加103.5g碳酸钾溶液;滴毕,搅拌反应10小时。反应毕,用110g稀盐酸调节ph=3~5;用300ml二氯甲烷分3次萃取,合并有机相,经水洗、干燥,减压浓缩,得23g培比洛芬,黄色油状物,纯度90%以上。加入50g混合溶剂,重复精制2~3次,得培比洛芬成品14g,类白色结晶粉末,m.p106~110℃,纯度99%以上。
- 还没有人留言评论。精彩留言会获得点赞!