一种UV交联单体及其制备方法与在制备含氟丙烯酸酯乳液中的应用
本发明属于丙烯酸酯乳液技术领域,具体涉及一种uv交联单体及其制备方法与在制备含氟丙烯酸酯乳液中的应用。
背景技术:
含氟聚合物具有许多应用,源于其具有比较特殊的元素和结构。从元素上看:自然界中,氟原子是除了氢原子以外最小的原子;另外,氟元素的电负性最大,c-f键能远远大于c-h键能,有良好的热稳定性;从结构上看:一方面,含氟聚合物的构象结构为螺旋结构,即含氟侧链很好的包裹着主链,形成氟屏蔽效应,使得主链不被破坏,另一方面,对称的链结构使得含氟聚合物表面为非极性。总体而言,含氟聚合物具有良好的化学惰性,这种化学惰性赋予了含氟聚合物较好的拒水、拒油、防污等功能。
含氟聚合物在整理剂上应用甚广,包括织物整理剂、纸张整理剂和皮革整理剂等。其中,应用最广泛的是织物整理剂,氟原子自身的低表面能,经过含氟织物整理剂处理后的织物,具有防水、防油、防污、抗紫外和阻燃等功能。
交联网状聚合物比线性聚合物具有更高的机械强度,如拉伸强度、冲击强度等;对于聚合物薄膜来说,网状聚合物具有更高的交联密度,使得薄膜的致密性较高,从而使原有的性能显著提高,如耐水性、抗渗透性和耐候性等。按照驱动交联的方式不同,提高交联度的方式可分为热交联、自交联和光交联。对于交联剂在织物整理剂上的应用,自交联的应用探索较多,如中国发明专利cn111662404a公开了一种自交联长氟碳丙烯酸酯聚合物乳液的制备方法,选择双丙酮丙烯酰胺等作为自交联剂;而对于光交联在织物整理剂上的应用探索则较少。而且该中国发明专利cn111662404a公开的自交联长氟碳丙烯酸酯聚合物乳液的制备方法,选择甲基丙烯酸十二氟庚酯等长碳链含氟单体,具有环境危害性和生物积累性,其次,选用阴离子乳化体系和非离子乳化体系复配,对基材附着力较差;最后,选用双丙酮丙烯酰胺等作为自交联剂,反应结束后还需要额外添加第二交联组分己二酸二酰肼,增加了工艺的复杂性。
技术实现要素:
为了克服现有技术存在的不足,本发明的目的是提供一种uv交联单体及其制备方法与在制备含氟丙烯酸酯乳液中的应用。
本发明提供了一种uv交联单体及其含氟丙烯酸酯共聚乳液的制备方法。所述uv交联单体可作为丙烯酸酯类单体参与聚合,也可以作为光交联剂来提高聚合物交联度;所述含氟丙烯酸酯共聚乳液利用所述uv交联单体与其他丙烯酸酯类单体通过乳液聚合制得,经uv光照后,乳胶膜交联度提升,吸水率下降,耐水性提高。
本发明先采用两步法合成一种可光交联单体,然后利用此单体与含氟丙烯酸酯类单体、其他丙烯酸酯类单体共聚,得到可uv交联的含氟丙烯酸酯乳液。
本发明的目的至少通过如下技术方案之一实现。
本发明提供的uv交联单体的制备方法,包括如下步骤:
(1)合成单溴取代二苯甲酮:将1,6-二溴己烷、4-羟基二苯甲酮、无水碳酸钾和有机溶剂混合,得到混合液a,在搅拌状态下进行遮光反应,得到反应液a,萃取,过滤取滤液,层析、旋蒸和干燥,得到单溴取代二苯甲酮;
(2)合成uv交联单体:将步骤(1)所述单溴取代二苯甲酮、不饱和酸、无水碳酸钾和有机溶剂混合均匀,得到混合液b,进行遮光反应,得到反应液b,萃取,过滤取滤液,层析、旋蒸和干燥,得到所述uv交联单体(粘稠状带有双键的可聚合二苯甲酮)。
进一步地,步骤(1)所述1,6-二溴己烷和4-羟基二苯甲酮的摩尔比为(2-5):1;
进一步地,步骤(1)所述4-羟基二苯甲酮与无水碳酸钾的摩尔比为1:(4-8);
进一步地,步骤(1)所述有机溶剂为丙酮、n,n-二甲基甲酰胺、二氯甲烷、氯仿、四氢呋喃、二甲基亚砜、甲苯、1,4-二氧六环中的一种以上;
进一步地,在步骤(1)所述混合液a中,有机溶剂的质量分数为10-50wt%;
进一步地,步骤(1)所述遮光反应的温度为50-65℃,遮光反应的时间为20-30h。
优选地,步骤(1)所述萃取使用的溶剂为二氯甲烷、n,n-二甲基甲酰胺、四氯甲烷中的一种。
优选地,步骤(1)所述层析采用的是石油醚与乙酸乙酯的混合液,所述石油醚与乙酸乙酯的体积比为8-10:1。
优选地,在所述混合液a中,有机溶剂的质量分数为20-40wt%。
进一步地,步骤(2)所述单溴取代二苯甲酮与不饱和酸的摩尔比为1:(1-10);
进一步地,步骤(2)所述不饱和酸为丙烯酸或甲基丙烯酸;
进一步地,步骤(2)所述单溴取代二苯甲酮与无水碳酸钾的摩尔比为1:(1-10);
进一步地,步骤(2)所述有机溶剂为丙酮、n,n-二甲基甲酰胺、二氯甲烷、氯仿、四氢呋喃、二甲基亚砜、甲苯、1,4-二氧六环中的一种以上;
进一步地,在步骤(2)所述混合液b中,有机溶剂的质量分数为10-50wt%;
进一步地,步骤(2)所述遮光反应的温度为50-65℃;
进一步地,步骤(2)所述遮光反应的时间为20-30h;
优选地,步骤(2)所述单溴取代二苯甲酮与所述不饱和酸的摩尔比为1:(2-5)。
优选地,步骤(2)所述单溴取代二苯甲酮与所述无水碳酸钾的摩尔比1:(4-8)。
优选地,步骤(2)所述萃取采用的溶剂为二氯甲烷、n,n-二甲基甲酰胺、四氯甲烷中的一种。
优选地,步骤(2)所述层析采用的是石油醚与乙酸乙酯的混合液,所述石油醚与乙酸乙酯的体积比为(8-10):1。
本发明提供一种由上述的制备方法制得的uv交联单体。
本发明提供的uv交联单体在制备可光固化含氟丙烯酸酯乳液中的应用,包括如下步骤:
(1)预乳化:将含氟丙烯酸酯单体、硬单体、软单体、功能单体和所述uv交联单体(光交联单体)混合均匀,得到混合单体;将非离子乳化剂和阳离子乳化剂溶于去离子水中,得到乳化剂水溶液;将所述混合单体滴加至所述乳化剂水溶液中,搅拌处理,得到预乳液;
(2)聚合:在反应釜中将步骤(1)所述预乳液分成两部分,标记为预乳液a和预乳液b;将非离子乳化剂、引发剂、助溶剂及去离子水混合,升温至75-85℃,然后在15-30分钟内滴加步骤(1)所述预乳液a,保温处理,得到混合液;往所述混合液中滴加引发剂溶液和预乳液b,升温进行加热处理,冷却至室温,出料,得到所述可光固化含氟丙烯酸酯乳液(含氟丙烯酸酯乳液)。
进一步地,步骤(1)所述含氟丙烯酸酯单体为全氟己基乙基丙烯酸酯或全氟己基乙基甲基丙烯酸酯;
进一步地,步骤(1)所述硬单体为(甲基)丙烯酸甲酯、甲基丙烯酸乙酯、苯乙烯、丙烯腈、甲基丙烯酸异冰片酯、甲基丙烯酸苄基酯中的一种以上;
进一步地,步骤(1)所述软单体为丙烯酸丁酯、丙烯酸异辛酯、(甲基)丙烯酸月桂酯、(甲基)丙烯酸十八酯、甲基丙烯酸-2-乙基己酯、甲基丙烯酸正辛酯中的一种以上;
进一步地,步骤(1)所述功能单体为(甲基)丙烯酸羟乙酯、(甲基)丙烯酸、(甲基)丙烯酸羟丙酯、甲基丙烯酸缩水甘油酯、n-羟甲基丙烯酰胺、n-丁氧基丙烯酰胺、二乙烯基苯、乙烯基三甲氧基硅烷、乙烯基三异丙氧基硅烷、γ-甲基丙烯酰氧丙基三甲氧基硅烷中的一种以上;
进一步地,在步骤(1)所述混合单体中,按照质量份数计,包括:
进一步地,步骤(1)所述所述非离子乳化剂为月桂醇聚氧乙烯醚-4(聚氧乙烯醚(4)月桂醇醚)、硬脂醇聚氧乙烯醚-2(聚氧乙烯(2)硬脂醇醚)、鲸蜡硬脂醇聚醚-20(鲸蜡硬脂醇聚醚-20)、硬脂醇聚氧乙烯醚-10(聚氧乙烯(10)硬脂醇醚)、硬脂醇聚氧乙烯醚-21(聚氧乙烯(21)硬脂醇醚)、硬脂醇聚氧乙烯醚-20(聚氧乙烯(20)硬脂醇醚)、烷基酚聚氧乙烯醚-20(聚氧乙烯(20)烷基酚醚)中的一种以上;
进一步地,步骤(1)所述阳离子乳化剂为十八烷基三甲基溴化铵、十八烷基三甲基氯化铵、十八烷基二甲基苄基氯化铵、十六烷基三甲基溴化铵、十六烷基三甲基氯化铵、gemini阳离子表面活性剂中的一种以上;
进一步地,步骤(1)所述非离子乳化剂与阳离子乳化剂的质量比为(2-4):1;所述非离子乳化剂与水的质量比为1:(8-12);
进一步地,步骤(1)所述乳化剂水溶液与混合单体的质量比为1:(2-3);
进一步地,步骤(1)所述搅拌处理的时间为20-30分钟。
进一步地,步骤(2)所述预乳液a的质量占预乳液质量的10-25wt%;
进一步地,步骤(2)所述预乳液a与非离子乳化剂的质量比为(12-16):1;
进一步地,步骤(2)所述非离子乳化剂为月桂醇聚氧乙烯醚-4(聚氧乙烯醚(4)月桂醇醚)、硬脂醇聚氧乙烯醚-2(聚氧乙烯(2)硬脂醇醚)、鲸蜡硬脂醇聚醚-20(鲸蜡硬脂醇聚醚-20)、硬脂醇聚氧乙烯醚-10(聚氧乙烯(10)硬脂醇醚)、硬脂醇聚氧乙烯醚-21(聚氧乙烯(21)硬脂醇醚)、硬脂醇聚氧乙烯醚-20(聚氧乙烯(20)硬脂醇醚)、烷基酚聚氧乙烯醚-20(聚氧乙烯(20)烷基酚醚)中的一种以上;
进一步地,步骤(2)所述引发剂为偶氮二异丁脒盐酸盐、偶氮二异丁咪唑啉盐酸盐、过硫酸钾-亚硫酸氢钠体系、过硫酸铵、过硫酸钾中的一种以上;所述助溶剂为异丙醇、三丙二醇、丙二醇乙醚、丙二醇甲醚、二乙二醇丁醚中的一种以上;
进一步地,步骤(2)所述非离子乳化剂与引发剂的质量比为1:(0.2-0.3);
进一步地,步骤(2)所述非离子乳化剂与助溶剂的质量比为1:(58-62);
进一步地,步骤(2)所述非离子乳化剂与水的质量比为1:(92-95)。
进一步地,步骤(2)所述保温处理的时间为15-20分钟;
进一步地,步骤(2)所述引发剂溶液为引发剂与水混合均匀的溶液;
进一步地,步骤(2)所述引发剂溶液的浓度为3-4wt%;
进一步地,步骤(2)所述非离子乳化剂与引发剂溶液的质量比为1:(1.2-1.4);所述引发剂溶液的滴加时间为2.5-4小时;
进一步地,步骤(2)所述预乳液b的滴加时间为2-3.5小时;
进一步地,步骤(2)所述加热处理的温度为90-95℃,加热处理的时间为1-2h。
优选地,所述含氟丙烯酸酯单体、硬单体、软单体、功能单体和光交联单体总质量占整个反应体系原料质量的25-35wt%。
为进一步实现实验目的,优选地,以占单体总质量百分比算,单体组分用量为:含氟丙烯酸酯单体占45-65%,硬单体占5-15%,软单体占5-30%,功能单体占1-10%,光交联单体占1-5%。
优选地,所述乳化剂总量占单体总量3-10wt%,优选地,阳离子乳化剂含量为1-5wt%。
优选地,所述助溶剂含量为占总溶剂含量的20-40wt%。
本发明提供一种由上述的应用方法制得的可光固化含氟丙烯酸酯乳液。
本发明提供的可光固化含氟丙烯酸酯乳液可以作为织物整理剂使用。
本发明使用该uv交联单体制备的可光固化含氟丙烯酸酯乳液,其粒径小,分布窄,热稳定性能良好,制备方法中所用含氟单体不含c8及以上的含氟烷基链,环境友好,应用于织物整理剂上,可以使织物耐水性能提高。
本发明通过1,6-二溴己烷、4-羟基二苯甲酮、甲基丙烯酸采用两步法合成可光交联单体,方法简单;使用有机溶剂易于获得,毒性不大,容易工业化生产;聚合物合成过程,采用乳液聚合方法可有效降低voc含量,使用全氟己基代替全氟辛基的含氟单体,避免全氟长烷基链(cnf2n+1-,n≥8)的生物积累性和毒性,满足欧盟的环境保护法;同时,使用光交联代替以往的热交联,对工业生产交联方式的改变具有指导意义;使用弱阳离子乳化体系,对于应用于织物整理剂具有良好的耐水性能。
与现有技术相比,本发明具有如下优点和有益效果:
(1)本发明制备的uv交联单体方法简单,实用性强;
(2)本发明提供的乳液制备方法中,使用的短碳链含氟丙烯酸酯单体(含氟碳数小于等于6),符合欧盟的环保要求,不会有生物积累性和毒性;
(3)本发明制备的乳液方法简单,自动滴加工艺保证反应速率平缓稳定,转化率高。
(4)本发明提供的乳液制备方法中,使用uv固化能源消耗减少,基材适用性高,交联固化后,织物的疏水性提高。
(5)本发明提供的乳液制备方法中,使用阳离子乳化剂代替部分非离子乳化剂复配成弱阳离子乳化体系,乳液聚合稳定好,而且对织物纤维具有有良好的附着力;
(6)本发明提供的乳液制备方法中,使用助溶剂代替部分去离子水,增加乳液干燥成膜的速度。
附图说明
图1为实施例1所得产物的核磁共振氢谱图。
图2为实施例2所得产物的核磁共振氢谱图。
图3为实施例3所得产物的核磁共振氢谱图。
图4为实施例4为所得产物得红外吸收光谱图。
图5为实施例5为所得产物得红外吸收光谱图。
图6为实施例6为所得产物得红外吸收光谱图。
图7为实施例7为所得产物得红外吸收光谱图。
具体实施方式
以下结合实例对本发明的具体实施作进一步说明,但本发明的实施和保护不限于此。需指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器未注明生产厂商者,视为可以通过市售购买得到的常规产品。
实施例1
一种uv交联单体通过以下方法制备得到:
1)待250ml四口烧瓶中升到55℃后,一次性加入41.77g的1,6-二溴己烷、12.22g的4-羟基二苯甲酮和51.12g无水碳酸钾,再加入60g丙酮,外设温度为55℃,在机械搅拌下,用锡纸遮光反应24小时,黄光完全褪去,出料,经二氯甲烷萃取、过滤取滤液、层析(层析使用的是石油醚与乙酸乙酯的混合液,石油醚与乙酸乙酯的体积比为8:1)、旋蒸和和干燥后,得到单溴取代二苯甲酮;
2)待250ml四口烧瓶中升到60℃后,一次性加入6.97g单溴取代二苯甲酮、4.77g甲基丙烯酸、15.33g无水碳酸钾,再加入60g的n,n-二甲基甲酰胺,用锡纸遮光反应24小时,出料,经二氯甲烷萃取、过滤取滤液、层析(层析使用的是石油醚与乙酸乙酯的混合液,石油醚与乙酸乙酯的体积比为8:1)、旋蒸和干燥后,得到uv交联单体。
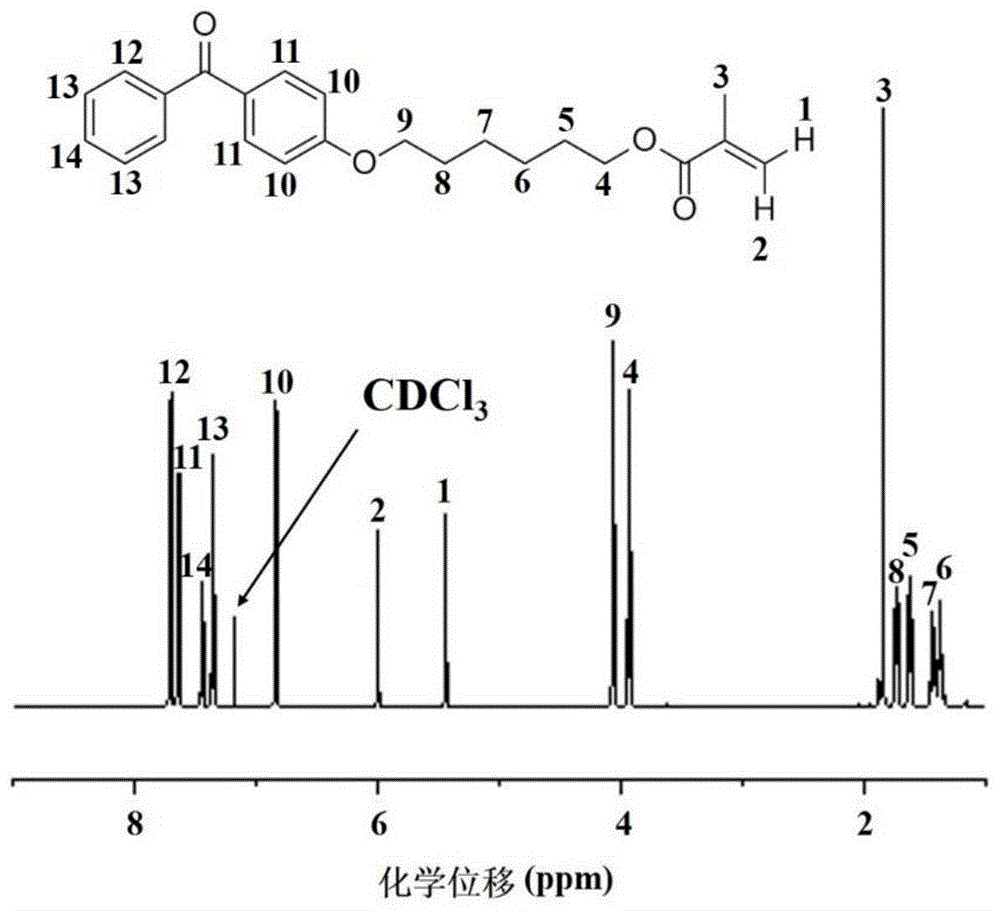
用氘代氯仿作为氘代试剂,测试uv交联单体的核磁共振氢谱,结果如图1所示,化学位移分析如下:1h-nmr(cdcl3tms)δ(ppm):7.81(d,2h,-arh-),7.75(d,2h,-arh-),7.56(t,1h,-arh-),7.47(t,2h,-arh-),6.84(d,2h,-arh-),6.04(s,1h,h-c=c),5.42(s,1h,h-c=c),4.10(t,2h,-ch2-o-ar),3.94(t,2h,-ch2-o-c=o),1.84(s,3h,-ch3-c=c),1.73(m,2h,-ch2-ch2-o-ar),1.64(m,2h,-ch2-ch2-o-c=o),1.42(m,2h,-ch2-ch2-ch2-o-ar),1.36(m,2h,-ch2-ch2-ch2-o-c=o)。
实施例2
一种uv交联单体通过以下方法制备得到:
1)待250ml四口烧瓶中升到60℃后,一次性加入30.08g的1,6-二溴己烷、12.22g的4-羟基二苯甲酮和51.12g无水碳酸钾,再加入60g丙酮,外设温度为55℃,在机械搅拌下,用锡纸遮光反应20小时,黄光完全褪去,出料,经二氯甲烷萃取、过滤取滤液、层析(层析使用的是石油醚与乙酸乙酯的混合液,石油醚与乙酸乙酯的体积比为9:1)、旋蒸和和干燥后,得到单溴取代二苯甲酮;
2)待250ml四口烧瓶中升到60℃后,一次性加入6.43g单溴取代二苯甲酮、4.40g甲基丙烯酸、14.14g无水碳酸钾,再加入60g的n,n-二甲基甲酰胺,用锡纸遮光反应20小时,出料,经二氯甲烷萃取、过滤取滤液、层析(层析使用的是石油醚与乙酸乙酯的混合液,石油醚与乙酸乙酯的体积比为9:1)、旋蒸和干燥后,得到uv交联单体。
用氘代氯仿作为氘代试剂,测试uv交联单体的核磁共振氢谱,结果如图2所示,化学位移分析如下:1h-nmr(cdcl3tms)δ(ppm):7.92(d,2h,-arh-),7.80(d,2h,-arh-),7.65(t,1h,-arh-),7.60(t,2h,-arh-),6.94(d,2h,-arh-),6.02(s,1h,h-c=c),5.40(s,1h,h-c=c),4.01(t,2h,-ch2-o-ar),3.89(t,2h,-ch2-o-c=o),1.80(s,3h,-ch3-c=c),1.71(m,2h,-ch2-ch2-o-ar),1.62(m,2h,-ch2-ch2-o-c=o),1.43(m,2h,-ch2-ch2-ch2-o-ar),1.33(m,2h,-ch2-ch2-ch2-o-c=o)。
实施例3
一种uv交联单体通过以下方法制备得到:
1)待250ml四口烧瓶中升到65℃后,一次性加入75g1,6-二溴己烷、12.22g4-羟基二苯甲酮和51.12g无水碳酸钾,再加入60g丙酮,外设温度为55℃,在机械搅拌下,用锡纸遮光反应24小时,黄光完全褪去,出料,经二氯甲烷萃取、过滤取滤液、层析(层析使用的是石油醚与乙酸乙酯的混合液,石油醚与乙酸乙酯的体积比为10:1)、旋蒸和和干燥后,得到单溴取代二苯甲酮;
2)待250ml四口烧瓶中升到60℃后,一次性加入6.74g单溴取代二苯甲酮、4.62g甲基丙烯酸、14.83g无水碳酸钾,再加入60g的n,n-二甲基甲酰胺,用锡纸遮光反应30小时,出料,经二氯甲烷萃取、过滤取滤液、层析(层析使用的是石油醚与乙酸乙酯的混合液,石油醚与乙酸乙酯的体积比为10:1)、旋蒸和干燥后,得到uv交联单体。
用氘代氯仿作为氘代试剂,测试uv交联单体的核磁共振氢谱,结果如图3所示,化学位移分析如下:1h-nmr(cdcl3tms)δ(ppm):7.71(d,2h,-arh-),7.64(d,2h,-arh-),7.60(t,1h,-arh-),7.47(t,2h,-arh-),6.90(d,2h,-arh-),6.01(s,1h,h-c=c),5.51(s,1h,h-c=c),4.15(t,2h,-ch2-o-ar),4.02(t,2h,-ch2-o-c=o),1.81(s,3h,-ch3-c=c),1.76(m,2h,-ch2-ch2-o-ar),1.61(m,2h,-ch2-ch2-o-c=o),1.45(m,2h,-ch2-ch2-ch2-o-ar),1.32(m,2h,-ch2-ch2-ch2-o-c=o)。
实施例4
一种可光固化含氟丙烯酸酯乳液(可uv交联含氟丙烯酸酯乳液)通过以下方法制备得到:
1)预乳化:将8.25g全氟己基乙基丙烯酸酯、1.70g甲基丙烯酸甲酯、4.3g甲基丙烯酸十八酯、0.3g甲基丙烯酸羟乙酯和0.45g的uv交联单体(实施例1制备的)混合均匀,得到混合单体,将0.6g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)和0.15g阳离子乳化剂(十八烷基三甲基溴化铵)溶于6g去离子水中,再将上述混合单体滴入上述乳化剂水溶液中,搅拌25分钟得到预乳液;
2)将14g去离子水、0.15g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)、0.045g引发剂(过硫酸铵)、9g助溶剂(二乙二醇丁醚)加入反应釜中,并升温至80℃,在15分钟内滴入2.3g预乳液,保温15分钟后,将0.21g引发剂(过硫酸铵)溶于6g去离子水中,配成引发剂水溶液;同时滴加剩余预乳液(18.7g)和上述引发剂水溶液,3.5小时内滴完,再升温至90℃,保温2小时,冷却至室温后出料得到可光固化含氟丙烯酸酯乳液。
将所得聚合物乳液用甲醇破乳、洗涤、干燥后,使用溴化钾压片法进行红外光谱测试,测试结果如图4所示。3432cm-1为羟基的对称伸缩振动,结合合成过程,证明有甲基丙烯酸羟乙酯参与聚合;3032cm-1为苯环上的碳氢对称伸缩振动,结合合成过程、交联度和吸水率测试,证明uv交联单体参与聚合;2919cm-1为-ch2-的反对称伸缩振动,2850cm-1为-ch2-的对称伸缩振动,是聚合物碳链和甲基丙烯酸十八酯聚合后侧链的十八烷基的吸收峰;1728cm-1为c=o的特征吸收峰,证明酯基的存在;1450cm-1为-ch3的特征吸收峰,为甲基丙烯酸甲酯的侧甲基的吸收峰;1076cm-1~1238cm-1为c-f、c-c等多种结构的特征吸收峰重叠后形成的谱带,结合合成过程和接触角测试结果,证明有全氟己基乙基丙烯酸酯单体(含氟单体)参与聚合。综上所述,在本实施案例4中,成功制备了可uv交联含氟丙烯酸酯乳液。
实施例5
1)预乳化:将8.25g全氟己基乙基丙烯酸酯、1.41g甲基丙烯酸甲酯、4.3g甲基丙烯酸十八酯、0.3g甲基丙烯酸羟乙酯和0.75g的uv交联单体(实施例2制备的)混合均匀,得到混合单体,将0.6g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)和0.15g阳离子乳化剂(十八烷基三甲基溴化铵)溶于6g去离子水中,再将上述混合单体滴入上述乳化剂水溶液中,搅拌20分钟得到预乳液;
2)将14g去离子水、0.15g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)、0.045g引发剂(过硫酸铵)、9g助溶剂(二乙二醇丁醚)加入反应釜中,并升温至80℃,在15分钟内滴入2.3g预乳液,保温15分钟后,将0.21g引发剂(过硫酸铵)溶于6g去离子水中,配成引发剂水溶液;同时滴加剩余预乳液(18.7g)和上述引发剂水溶液,3.5小时内滴完,再升温至90℃,保温2小时,冷却至室温后出料得到可光固化含氟丙烯酸酯乳液。
将所得聚合物乳液用甲醇破乳、洗涤、干燥后,使用溴化钾压片法进行红外光谱测试,测试结果如图5所示。3435cm-1为羟基的对称伸缩振动,结合合成过程,证明有甲基丙烯酸羟乙酯参与聚合;3020cm-1为苯环上的碳氢对称伸缩振动,结合合成过程、交联度和吸水率测试,证明uv交联单体参与聚合;2915cm-1为-ch2-的反对称伸缩振动,2865cm-1为-ch2-的对称伸缩振动,是聚合物碳链和甲基丙烯酸十八酯聚合后侧链的十八烷基的吸收峰;1724cm-1为c=o的特征吸收峰,证明酯基的存在;1469cm-1为-ch3的特征吸收峰,为甲基丙烯酸甲酯的侧甲基的吸收峰;1137cm-1~1234cm-1为c-f、c-c等多种结构的特征吸收峰重叠后形成的谱带,结合合成过程和接触角测试结果,证明有全氟己基乙基丙烯酸酯单体参与聚合。综上所述,在本实施案例5中,成功制备了可uv交联含氟丙烯酸酯乳液。
对比例1
1)预乳化:将8.25g全氟己基乙基丙烯酸酯、2.15g甲基丙烯酸甲酯、4.3g甲基丙烯酸十八酯、0.3g甲基丙烯酸羟乙酯混合均匀,得到混合单体,将0.6g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)和0.15g阳离子乳化剂(十八烷基三甲基溴化铵)溶于6g去离子水中,再将上述混合单体滴入上述乳化剂水溶液中,搅拌30分钟得到预乳液;
2)将14g去离子水、0.15g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)、0.045g引发剂(过硫酸铵)、9g助溶剂(二乙二醇丁醚)加入反应釜中,并升温至80℃,在15分钟内滴入2.3g预乳液,保温15分钟后,将0.21g引发剂(过硫酸铵)溶于6g去离子水中,配成引发剂水溶液;同时滴加剩余预乳液(18.7g)和上述引发剂水溶液,3.5小时内滴完,再升温至90℃,保温2小时,冷却至室温后出料得到含氟丙烯酸酯乳液。
将所得聚合物乳液用甲醇破乳、洗涤、干燥后,使用溴化钾压片法进行红外光谱测试,测试结果如图6所示。3486cm-1为羟基的对称伸缩振动,结合合成过程,证明有甲基丙烯酸羟乙酯参与聚合;2917cm-1为-ch2-的反对称伸缩振动,2873cm-1为-ch2-的对称伸缩振动,是聚合物碳链和甲基丙烯酸十八酯聚合后侧链的十八烷基的吸收峰;1735cm-1为c=o的特征吸收峰,证明酯基的存在;1469cm-1为-ch3的特征吸收峰,为甲基丙烯酸甲酯的侧甲基的吸收峰;1079cm-1230cm-1为c-f、c-c等多种结构的特征吸收峰重叠后形成的谱带,结合合成过程和接触角测试结果,证明有全氟己基乙基丙烯酸酯单体参与聚合。综上所述,在对比例1中,成功制备了含氟丙烯酸酯乳液。
对比例2
1)预乳化:将8.25g全氟己基乙基丙烯酸酯、1.41g甲基丙烯酸甲酯、5.06g甲基丙烯酸十八酯、0.3g甲基丙烯酸羟乙酯混合均匀,得到混合单体,将0.6g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)和0.15g阳离子乳化剂(十八烷基三甲基溴化铵)溶于6g去离子水中,再将上述混合单体滴入上述乳化剂水溶液中,搅拌20-30分钟得到预乳液;
2)将14g去离子水、0.15g非离子乳化剂(聚氧乙烯醚(4)月桂醇醚)、0.045g引发剂(过硫酸铵)、9g助溶剂(二乙二醇丁醚)加入反应釜中,并升温至80℃,在15分钟内滴入2.3g预乳液,保温15分钟后,将0.21g引发剂(过硫酸铵)溶于6g去离子水中,配成引发剂水溶液;同时滴加剩余预乳液(18.7g)和上述引发剂水溶液,3.5小时内滴完,再升温至90℃,保温2小时,冷却至室温后出料得到含氟丙烯酸酯乳液。
将所得聚合物乳液用甲醇破乳、洗涤、干燥后,使用溴化钾压片法进行红外光谱测试,测试结果如图7所示。3479cm-1为羟基的对称伸缩振动,结合合成过程,证明有甲基丙烯酸羟乙酯参与聚合;2931cm-1为-ch2-的反对称伸缩振动,2865cm-1为-ch2-的对称伸缩振动,是聚合物碳链和甲基丙烯酸十八酯聚合后侧链的十八烷基的吸收峰;1735cm-1为c=o的特征吸收峰,证明酯基的存在;1485cm-1为-ch3的特征吸收峰,为甲基丙烯酸甲酯的侧甲基的吸收峰;1087cm-1~1238cm-1为c-f、c-c等多种结构的特征吸收峰重叠后形成的谱带,结合合成过程和接触角测试结果,证明有全氟己基乙基丙烯酸酯单体参与聚合。综上所述,在本对比例2中,成功制备了含氟丙烯酸酯乳液。
将各实施例聚合物乳液、对比例的聚合物乳液分别用自来水稀释至固含量为千分之一,将伞布春亚纺织物浸渍于稀释后的乳液中,轧干,乳液轧余率为70-80%,然后用紫外辐射光固化机光照,光照条件为:紫外辐射距离10cm,辐射强度为36mw/cm2,时间为600秒,光照结束后在180℃中烘烤45秒,得到处理后织物。
使用索氏提取器测试织物交联度,测试条件为:四氢呋喃作为溶剂,提取24小时,参照标准gb/t24368-2009测试织物经索氏提取前后的接触角,参照标准gb/t21655.1-2008测试织物的吸水率。各项测试结果如表1所示。
由表1各项测试结果可知,引入uv交联单体的含氟丙烯酸酯乳液(实施例4和实施例5),经uv交联后,织物的交联度提升、吸水率下降,经过索氏提取后,织物的接触角变化不大;对比不含uv交联单体的含氟丙烯酸酯乳液(对比例1和对比例2),经过索氏提取后,织物的接触角下降幅度较大。
表1各实施例和对比例的测试结果数据表
本发明实施例采用两步法合成uv交联单体,合成工艺简单,溶剂易于获得;利用上述uv交联单体,本发明实施例采用预乳化半连续种子乳液的方法合成可uv交联含氟丙烯酸酯乳液,使用全氟己基代替全氟辛基的含氟单体具有环境友好性;通过上述测试结果可知,经uv交联后,织物的耐水性能提升;uv交联对基材的普适性,具有一定的应用前景。
以上实施例仅为本发明较优的实施方式,仅用于解释本发明,而非限制本发明,本领域技术人员在未脱离本发明精神实质下所作的改变、替换、修饰等均应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!