α-酸衍生物及其制备方法和用途
本申请是申请号为201610944141.8、申请日为2016年11月2日、发明名称“α-酸衍生物及其制备方法和用途”的分案申请。
本发明属于医药技术领域,具体涉及新的α-酸衍生物及其制备方法和应用。
背景技术:
啤酒花(humuluslupulusl.)又称忽布,香蛇麻,性味甘苦,微平。是桑科葎草属多年生草质蔓生藤本,雌雄异株,花单性,雌性球穗花序简称酒花。它是一种较耐寒不耐热的植物,主要分布于我国西北地区、新疆北部、东北、华东及山东、甘肃、陕西等地。目前,我国啤酒花产量约占全世界的13%,仅次于美国和德国,居世界第三位。
啤酒花是酿造啤酒的主要原料之一,它能够赋予啤酒特殊的苦味和独特的风味,增强啤酒的非生物稳定性和泡持性,并具有一定的防腐性能,被誉为“啤酒的灵魂”。除主要用于啤酒酿造外,啤酒花作为一种重要的药用植物,它的应用历史也很悠久。13世纪开始,啤酒花开始作为草药来使用。我国明代李时珍的《本草纲目》所引的宋代《开宝本草》中的一种中药“陀得花”指的就是啤酒花。据报道,啤酒花可用于防腐、健胃、安神、镇定、催眠、杀菌、抗炎和利尿,还可用于治疗消化不良、失眠、肺结核、麻风病等。
近年来对啤酒花提取物或其中所含有的化合物的药理作用多集中在抗菌、抗氧化、镇静和雌激素样作用等方面。啤酒花中化学成分多样,以黄酮类、树脂类(α-酸类和β-酸类)及挥发油类化合物为其化学特性,其中树脂类成分多具有显著的生物活性,是啤酒花药效成分的主要代表。由于α-酸类成分极性差异较小、立体化学问题复杂,因而在分离纯化、结构鉴定和评价单体化合物药理活性、新药开发过程中有一定困难。
本专利提供了一种从啤酒花中制备新α-酸衍生物的方法,并阐明了其在保肝和抗神经炎症方面的医药用途。
技术实现要素:
本发明的目的在于提供新α-酸衍生物及其制备方法和新的医药用途。
本发明提供了新α-酸衍生物,其具有如下结构。
r1为c1-c4烷基,r2,r3,r4为氢或c1-c4烷基;
优选地,r1为甲基,乙基,丙基,异丙基,正丁基,2-甲基丙基,1,1-二甲基乙基;
r2,r3,r4为甲基,乙基,丙基,异丙基,正丁基,2-甲基丙基,1,1-二甲基乙基。
进一步地,本发明优选如下衍生物:
本发明还提供了所述新的α-酸类衍生物1-14的制备方法,该方法包括如下步骤:
(1)啤酒花(humuluslupulusl.)的雌花序用乙醇提取,回收提取液得粗提物;
(2)步骤(1)所得粗提物经硅胶柱色谱法分离,以石油醚和乙酸乙酯混合溶剂梯度洗脱、或以石油醚和丙酮混合溶剂、或氯仿和丙酮混合溶剂、或二氯甲烷和丙酮、或氯仿和甲醇、或二氯甲烷和甲醇的混合溶剂梯度洗脱;
(3)上述步骤(2)中所得流分100:1~100:25经ods色谱分离,以甲醇和水混合溶剂为流动相梯度洗脱,或以乙腈和水混合溶剂为流动相梯度洗脱;
(4)上述步骤(3)中所得流分1:9~7:3经hplc-rid10a进一步分离,以甲醇和水混合溶剂为流动相梯度洗脱,或以乙腈和水混合溶剂为流动相梯度洗脱,得到α-酸类1-7,9和10的消旋混合物,11和12的消旋混合物。
(5)上述步骤(3)所得流分1:9~7:3经反复重结晶技术得到新的α-酸衍生物8,以及13和14的消旋混合物。
(6)上述步骤(4)和(5)所得的新α-酸衍生物9和10的消旋混合物,11和12的消旋混合物,13和14的消旋混合物经hplc手性拆分,以正己烷和乙醇混合溶剂为拆分溶剂得到新的α-酸衍生物9,10,11,12,13,14。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,所述啤酒花为桑科(moraceae)葎草属(humulus)植物啤酒花的雌花序。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,步骤(1)中所述提取方法为加热回流提取或加热超声提取1~3次,所用溶剂为50%~100%的乙醇,药材:溶剂的重量体积比为1:5~1:25。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,步骤(2)中所述石油醚和乙酸乙酯混合溶剂的比例为100:1~1:1,优选100:6~4:1,或石油醚和丙酮混合溶剂的比例为100:1~1:1,优选100:3~100:5,或氯仿和丙酮、或二氯甲烷和丙酮混合溶剂的比例为100:1~100:20,优选100:2~100:15、或氯仿和甲醇、或二氯甲烷和甲醇的混合溶剂梯度洗脱混合溶剂的比例为100:1~100:20,优选100:1~10:1。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,步骤(3)中所述甲醇和水混合溶剂,混合溶剂的比例为2:8~9:1,优选4:6~7:3,或乙腈和水混合溶剂,混合溶剂的比例为1:9~7:3,优选1:9~1:1。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,步骤(4)中所述流动相甲醇和水混合溶剂的比例为1:1~8:2,优选6:4~7:3,乙腈和水混合溶剂的比例为3:7~1:1,优选4:6~1:1。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,步骤(5)中甲醇和水混合溶剂的体积比例为:6:4~9:1,优选6:4~8:2,乙腈和水混合溶剂的体积比例为2:8~7:3,优选2:8~4:6。
本发明提供的所述新的α-酸类衍生物1-14的制备方法,步骤(6)中hplc手性柱拆分所应用洗脱剂为正己烷和乙醇的混合溶剂的体积比例为:90:10~95:5,优选93:7~95:5。
本发明以h2o2诱导hepg2和l-02肝细胞氧化损伤模型和lps诱导的bv2小胶质细胞过度活化模型,对制备得到的新的α-酸衍生物1-14的保肝及抗神经炎症活性进行了评价。结果显示,新化合物2,8,9,10,11,12,13,14能够显著的保护h2o2引起的hepg2和l-02肝细胞氧化损伤,化合物7具有中等强度的肝细胞保护活性。新α-酸类衍生物1,9,10,11,12,13,14(30μm,100μm),3,5,7(100μm)能够显著的抑制lps诱导的过度活化的bv2小胶质细胞no的释放,表现出显著的抗神经炎症活性。因此,本发明中制备的新α-酸类化合物可在开发保肝和治疗神经退行性疾病药物方面应用。
本发有首次提供了以啤酒花为原料,制备、鉴定14个新α-酸衍生物的方法,并系统评价了其肝保护、神经保护方面的活性,阐明了其在开发肝保护和治疗神经退行性疾病药物方面的应用。
附图说明
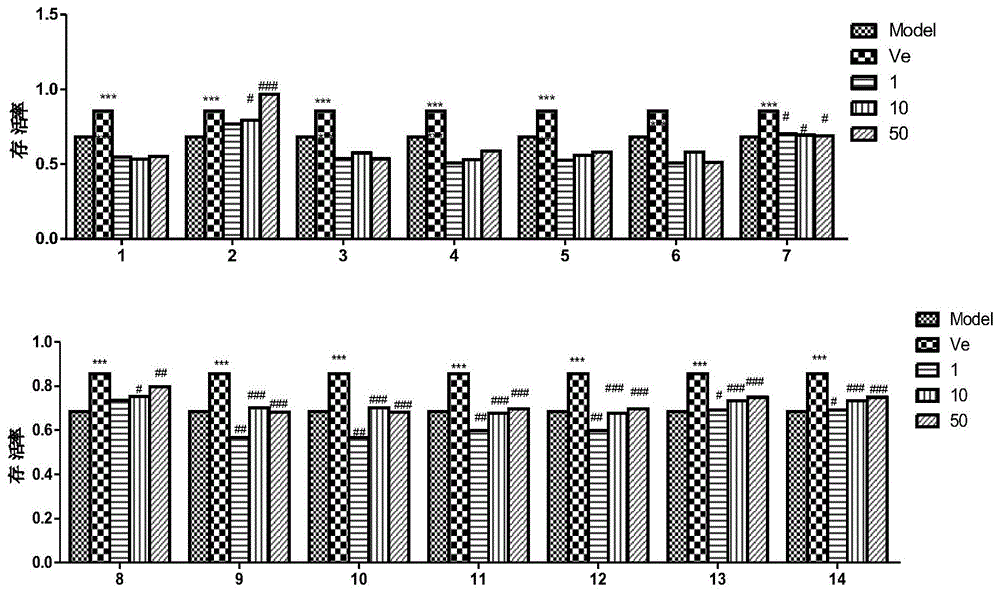
图1为不同化合物对h2o2损伤hepg2存活率的影响;
图2为不同化合物对h2o2损伤的hepg2细胞的保护率;
图3为不同化合物对h2o2损伤的l-02存活率的影响;
图4为不同化合物对h2o2损伤的l-02细胞的保护率。
具体实施方式
下面的实施例将对本发明予以进一步的说明,但并不因此而限制本发明。
实施例1
(1)啤酒花900g用50%的乙醇加热回流提取1次(用量:22.5l),减压回收提取液得粗提物;
(2)步骤(1)所得乙醇提取物经硅胶柱色谱法分离,依次以石油醚:乙酸乙酯100:1,100:3,100:6,100:8,8:1,7:1,2:1,1:1洗脱;
(3)上述步骤(2)中所得的石油醚:乙酸乙酯100:6流分经ods色谱分离,以甲醇/水2:8,4:6,1:1,6:4,9:1为流动相梯度洗脱;
(4)上述步骤(3)中所得甲醇:水(6:4)流分经hplc-rid色谱分离,以甲醇:水(7:3)为流动相洗脱得到新α-酸,化合物9和10的消旋混合物(tr=45min,收率0.012%),化合物4(tr=60min)(收率0.048%)。化合物9和10的消旋混合物再经手性柱色谱分离,以正己烷:乙醇(95:5)为流动相洗脱得到新的α-酸9(10.565min),10(12.826min)(收率各为0.006%)。
(5)上述步骤(3)中所得甲醇:水(1:1)流分经结晶溶剂甲醇:水(9:1),进行反复重结晶技术得化合物13和14的消旋混合物(收率0.043%)。化合物13和14的消旋混合物再经手性柱色谱分离,以正己烷:乙醇(95:5)为流动相洗脱得到新的α-酸13(7.220min),14(9.597min)(收率各为0.021%)。
(6)上述步骤(2)中所得的石油醚:乙酸乙酯100:8流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱。
(7)上述步骤(6)中所得甲醇:水(1:1)流分经hplc-rid色谱分离,以甲醇:水(7:3)为流动相洗脱得到新α-酸,化合物1(tr=50min)(收率0.077%)和化合物5(tr=64min)(收率0.099%)。
(8)上述步骤(2)中所得的石油醚:乙酸乙酯8:1流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱;
(9)上述步骤(8)中所得甲醇:水(1:1)流分经hplc-rid色谱分离,以甲醇:水(8:2)为流动相洗脱得到新α-酸,化合物7(tr=60min)(收率0.028%)。
(10)上述步骤(2)中所得的石油醚:乙酸乙酯7:1流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱;
(11)上述步骤(10)中所得甲醇:水(6:4)流分经hplc-rid色谱分离,以甲醇:水(72:28)为流动相洗脱得到新α-酸,化合物2(tr=48min,收率0.016%),化合物3(tr=59min,收率0.042%),化合物6(tr=35min,收率0.017%),化合物11和12的消旋混合物(tr=65min,收率0.010%)。化合物11和12的消旋混合物再经手性柱色谱分离,以正己烷:乙醇(95:5)为流动相洗脱得到新的α-酸11(9.958min),12(11.304min)(收率各为0.005%)。
(12)上述步骤(2)中所得的石油醚:乙酸乙酯4:1流分经ods柱色谱分离,以甲醇/水2:8,4:6,1:1,6:4,9:1为流动相梯度洗脱;
(13)上述步骤(12)中所得甲醇:水(1:1)流分经结晶溶剂甲醇:水(8:2),进行反复重结晶技术得新α-酸,化合物8(收率0.043%)。
根据化合物1-14的理化性质和波谱数据鉴定了其结构。
化合物1:(3r,8s)-humulonea的结构鉴定数据如下:
淡黄色油状固体(二氯甲烷),
各个片段的连接方式由hmbc确定。在hmbc谱中,δh2.83(1h,dd,j=13.8,6.6hz,h-2"a),2.79(1h,dd,j=13.8,7.4hz,h-2"b)与200.9(c-1")远程相关,所以异丁基与羰基相连;由δh18.84(5-oh)与200.9(c-1")相关,得异戊酰基与α-acids母核直接相连;δh2.59(1h,dd,j=13.8,6.6hz,h-1”'a),2.50(1h,dd,j=13.8,7.4hz,h-1”'b)与77.5(c-5),167.4(c-8a),195.5(c-7)远程相关,进一步说明了α-acids4位异戊烯基与5位羟基环合成吡喃环。δh3.81(1h,t,j=4.7hz,h-3)与101.7(c-4a)相关,δh2.56(2h,d,j=4.7hz,h-4)与68.3(c-3),81.8(c-2),101.7(c-4a)相关,由此可确定吡喃环的存在。
化合物1的相对构型借助noesy来确定。在noesy谱中,δh3.81(h-3)与2.56(h-4),1.37(2'-ch3)相关;δh1.43(2'-ch3)与5.01(h-2”')、1.66(4”'-ch3)相关;由此可推测异戊烯基与1'-ch3在同侧,与2'-ch3在异侧。
该化合物的绝对构型通过运用tddft(时密度泛函理论)方法进行ecd计算确定,将实验测出的谱图与计算得到的对映异构体为(3r,8s)的ecd图谱进行比较,谱图相吻合,表现为在205-210nm和280-320nm区域有正康顿效应(cottoneffect),在220-260nm和345-400nm区域有负康顿效应。因此,确定该化合物的绝对构型为3r,8s。
表1化合物1的nmr数据归属
化合物2:(3s,8s)-humulonea的结构鉴定数据如下:
淡黄色油状固体(二氯甲烷),
各个片段的连接方式由hmbc谱确定。在hmbc谱中,δh2.86-2.77(2h,m,h-2")与200.7(c-1")远程相关,所以异丁基与羰基相连;δh18.79(5-oh)与200.7(c-1")相关,提示异戊酰基与α-acids母核直接相连;δh2.58(1h,dd,j=13.8,7.8hz,h-1”'a),2.46(1h,dd,j=13.8,7.6hz,h-1”'b)与77.6(c-8),167.2(c-8a),195.5(c-7)远程相关,进一步说明了α-acids4位异戊烯基与5位羟基环合成吡喃环。δh3.76(1h,t,j=5.4hz,h-3)与81.6(c-2),102.2(c-4a)相关,δh2.74(1h,dd,j=17.5,5.0hz,h-4a),2.35(1h,dd,17.5,5.9hz,h-4b)与69.0(c-3),81.6(c-2),102.2(c-4a)相关,由此可确定吡喃环的存在。
化合物2的相对构型借助noesy谱来确定。在noesy谱中,3.76(h-3)与2.74(h-4a),4.95(h-2”'),1.37(2'-ch3)相关;2.35(h-4b)与1.42(1'-ch3)相关;2.74(h-4a)与3.76(h-3),1.37(2'-ch3)相关。可知异戊烯基与h-3,2'-ch3在同侧,与1'-ch3在异侧。
该化合物的绝对构型是通过其cd光谱与化合物1的cd光谱进行比较确定的,二者在260-380之间均为负康顿效应(cottoneffect),这是由α、β不饱和酮基团引起的,由此可确定化合物2的8位与化合物1的8位构型相同,均为s构型。两个化合物cd光谱的差异在于190-200nm波段线型不同,这可能是由于化合物1与化合物2的3位绝对构型不同造成的,故化合物2的绝对构型初步定为(3s,8s),又因为化合物2的3位绝对构型可由noe进一步确定为3s构型,进而证实了ecd计算结果。因此,确定该化合物的绝对构型为3s,8s。
表2化合物2的nmr数据归属
化合物3:(3s,8s)-cohumulonea的结构鉴定数据如下:
淡黄色油状物(二氯甲烷),
各个片段的连接方式由hmbc谱确定。在hmbc谱中,δh3.74(1h,m,h-2")与205.7(c-1")远程相关,所以异丙基与羰基相连;由δh18.92(5-oh)与205.7(c-1")相关,则异丁酰基与α-acids母核直接相连;δh2.59(1h,dd,j=13.8,7.7hz,h-1”'a),2.46(1h,dd,j=13.8,7.9hz,h-1”'b)与77.7(c-8),167.2(c-8a),195.4(c-7)远程相关,进一步说明了α-acids4位异戊烯基与5位羟基环合成吡喃环。δh3.76(1h,m,h-3)与81.6(c-2),102.0(c-4a)相关,δh2.71(1h,dd,j=17.5,4.9hz,h-4a),2.36(1h,dd,j=17.5,5.7hz,h-4b)与69.0(c-3),81.6(c-2),102.0(c-4a)相关,由此可确定吡喃环的存在。
化合物3的相对构型借助noesy谱来确定。在noesy谱中,3.76(h-3)与2.71(h-4a),4.96(h-2”'),1.38(2'-ch3)相关;2.36(h-4b)与1.42(1'-ch3),相关;2.71(h-4a)与3.76(h-3),1.38(2'-ch3)相关,可知异戊烯基与h-3,2'-ch3在同侧,与1'-ch3在异侧。
该化合物的绝对构型是通过其cd光谱与化合物2的cd光谱进行比较确定的,二者均在195-215nm和260-330nm区域有正康顿效应(cottoneffect),在220-260nm和350-400nm区域有负康顿效应,并且cd光谱在195-215nm之间线型相近,故初步确定化合物3的绝对构型为(3s,8s)。又因为化合物3与化合物2的cd谱相比,二者在260-380之间的cotton效应相同,这是由α、β不饱和酮基团引起的,由此可确定化合物3的8位与化合物2的8位构型相同,均为s构型,化合物3的3位绝对构型可由noe进一步确定为3s构型,进而证实了ecd计算结果,故最终确定化合物3的绝对构型为(3s,8s)。
表3化合物3的nmr数据归属
化合物4:(2s,7s)-humuloneb的结构鉴定数据如下:
红褐色油状物(二氯甲烷),
各个片段的连接方式由hmbc谱确定。在hmbc谱中,δh2.82(1h,dd,j=13.8,6.8hz),2.76(1h,dd,j=13.8,7.5hz)与199.7(c-1")远程相关,所以异丁基与羰基相连;δh18.50(4-oh)与199.7(c-1")相关,提示异戊酰基与α-acids母核直接相连;δh2.62(1h,dd,j=14.1,8.7hz,h-2"a),2.43(1h,dd,j=14.1,5.7hz,h-2"b)与δc76.2(c-7),173.1(c-7a),196.1(c-6)远程相关,进一步说明了α-acids4位异戊烯基与5位羟基环合成五元环。
化合物4的相对构型借助noesy谱进行了确定,在noesy谱中:4.84(h-2)与2.90(h-3b)强相关,与3.00(h-3a)弱相关;5.23(h-2”')与1.37(2'-ch3)有相关,与4.84(h-2)无相关,可知h-2与h-3b同侧,h-3a与2位叔丙醇、7位异戊烯基同侧。
该化合物的绝对构型通过运用tddft(时密度泛函理论)方法进行ecd计算确定,将实验测出的谱图与计算得到的(2s,7s)的ecd图谱进行比较,二者谱图相吻合,表现为在190-200nm和260-325nm区域有正康顿效应(cottoneffect),在210-250nm和330-370nm区域有负康顿效应。因此,确定该化合物的绝对构型为(2s,7s)。
表4化合物4的nmr数据归属
化合物5:(2r,7s)-humuloneb的结构鉴定数据如下:
红褐色油状物(二氯甲烷),
各个片段的连接方式由hmbc谱确定。在hmbc谱中,δh2.81(1h,dd,j=13.7,7.5hz,h-2"a),2.76(1h,dd,j=13.7,6.7hz,h-2"b)与199.3(c-1")远程相关,所以异丁基与羰基相连;δh18.47(4-oh)与199.3(c-1")相关,提示异戊酰基与α-acids母核直接相连;δh2.56(2h,dd,j=7.5,4.6hz,h-2")与δc75.9(c-7),172.8(c-7a),196.0(c-6)远程相关,进一步说明了α-acids4位异戊烯基与5位羟基环合成五元呋喃环。
化合物5的相对构型借助noesy谱来确定,在noesy谱中4.76(h-2)与2.93(h-3a)强相关,与2.88(h-3b)弱相关;2.88(h-3b)与1.37(2'-ch3),1.17(3'-ch3)有相关,1.65(4”'-ch3)与1.37(2'-ch3),1.17(3'-ch3),有相关,可知h-2与h-3a、7位异戊烯基同侧同侧,h-3b与2位叔丙醇同侧。
化合物5的绝对构型是通过与化合物4的实测cd光谱及ecd计算光谱进行比较确定的,二者在260-380之间的cotton效应相同,这是由α、β不饱和酮键的n-π*跃迁引起的,由此可确定化合物5的7位与化合物4的7位构型相同,均为s构型,二者的cd光谱的区别在于波长范围在190-210nm之间虽都为正cotton效应,但cd曲线线型走势不同,这是由2位绝对构型相反造成的cd曲线差异,故化合物5的2位绝对构型为2r。又化合物5的2位构型也可由noesy谱进一步确定为2r,因此,确定该化合物的绝对构型为(2r,7s)。
表5化合物5的nmr数据归属
化合物6:(2r,7s)-cohumuloneb的结构鉴定数据如下:
红褐色油状物(二氯甲烷),
各个片段的连接方式由hmbc谱确定。在hmbc谱中,δh3.74(1h,m)与204.7远程相关,所以异丙基与羰基相连;δh18.68与204.7(c-1")相关,提示异丁酰基与α-acids母核直接相连;δh2.63(1h,dd,j=14.5,8.6hz,h-2"a),2.43(1h,dd,j=14.5,5.1hz,h-2"b)与76.3(c-7),173.3(c-7a),196.0(c-6)远程相关,进一步说明了α-酸4位异戊烯基与5位羟基环合成五元呋喃环。
化合物6的相对构型由noesy谱来确定,在noesy谱中,由δh4.84(h-2)与2.91(h-3b)强相关,与3.00(h-3a)弱相关;δh5.24(h-2”')与1.37(2'-ch3)有相关,与4.84(h-2)无相关,可知h-2与h-3b同侧,h-3a与2位叔丙醇、7位异戊烯基同侧。
化合物6的绝对构型是通过将其实测cd谱图与化合物4计算得到的(2s,7s)的ecd图谱进行比较确定的,二者在260-380之间的cotton效应相同,这是由α、β不饱和酮键的n-π*跃迁引起的,由此可确定化合物6的7位与化合物4的7位构型相同,均为s构型,化合物6的2位构型可由noesy谱进一步确定为2s构型。因此,确定该化合物的绝对构型为(2s,7s)。
表6化合物6的nmr数据归属
化合物7:(2s,3as)-humulonec的结构鉴定数据如下:
淡黄色油状物(二氯甲烷),
各个片段的连接方式由hmbc谱确定。在hmbc谱中,δh2.84(1h,dd,j=13.5,6.4hz,h-2"a),2.74(1h,dd,j=13.5,7.5hz,h-2"b)与199.4(c-1")远程相关,所以异丁基与羰基相连;δh18.81(6-oh)与199.4(c-1")相关,提示异戊酰基与α-acids母核直接相连;δh3.09(1h,dd,j=14.5,6.6hz,h-1”'a),3.03(1h,dd,j=14.5,8.3hz,h-1”'b)与δc108.7(c-7),120.9(c-2'"),133.0(c-3'"),170.4(c-7a),193.2(c-6)远程相关,进一步说明了α-酸6位异戊烯基与5位羟基环合成五元呋喃环。
化合物7的相对构型通过noesy谱不能确定,两个手性碳共有四种相对构型。
该化合物的绝对构型通过运用tddft(时密度泛函理论)方法进行ecd计算确定,将实验测出的谱图与计算得到的对映异构体(2s,3as),(2s,3ar),(2r,3ar),(2s,3as)的ecd图谱进行比较,与计算谱图(2s,3as)相吻合,表现为在190-225nm和260-340nm区域有负康顿效应(cottoneffect),在245-275nm和340-400nm区域有正康顿效应。因此,确定该化合物的绝对构型为(2s,3as)。
表7化合物7的nmr数据归属
化合物8:(3s,3'r,5r)-humuloneacidb的结构鉴定数据如下:
无色针状结晶(甲醇),
各个片段的连接方式由hmbc谱确定,hmbc谱显示:δh2.96(1h,t-like,j=7.4hz,h-4')与24.3(3"-ch3),29.5(2"-ch3),31.6(c-4'),35.5(c-4),58.9(c-3),88.2(c-2'),173.0(c-6),175.0(c-5')相关,提示a和b环通过c-3、3'直接相连;δh4.67(1h,dd,j=9.9,6.7hz,h-5)与24.8(3'"-ch3),26.1(2'"-ch3),35.5(c-4)相关,提示异丙醇基与c-5相连。
化合物8的相对构型借助noesy谱进行了确证,在noesy谱中,4.67(h-5)与2.65(h-4a)相关;2.55(h-4b)与2.96(h-3'),1.17(3'"-ch3)相关;2.96(h-3')与1.57(1"-ch3),2.55(h-4b),2.72(h-4'b)相关。由此可得h-5与h-4a,2"-ch3,h-4'b在同侧;h-4b与h-4'a,3'"-ch3,h-3'在同侧,又羧基氢为活泼氢,故其相对构型有四种。
该化合物的绝对构型通过运用tddft方法进行ecd计算确定。将实验测出的谱图与计算得到的对映异构体(3s,3'r,5r),(3r,3's,5s)的ecd图谱进行比较,与计算谱图(3s,3'r,5r)相吻合,表现为在190-250nm区域有正康顿效应(cottoneffect)。由此确定化合物8的绝对构型为3s,3'r,5r。
表8化合物8的nmr数据归属
化合物9和10消旋体:lupuloneh的结构鉴定数据如下:
无色针状结晶(甲醇),
各个片段的连接方式由hmbc谱来确定,δh2.99(1h,dd,j=15.5,6.8hz,h-2"a),2.86(1h,dd,j=15.5,6.9hz,h-2"b)与δc205.9(c-1"),162.9(c-7a)相关,则异丁基与羰基碳相连,且异戊酰基连在7位碳上;δh7.19(1h,d,j=8.2hz,h-4)与29.5(c-3)相关,δh3.15(1h,dd,j=15.3,9.1hz,h-3a),3.07(1h,dd,j=15.1,9.6hz,h-3b)与71.7(c-1'),91.6(c-2),117.6(c-3a)相关,提示五元呋喃环氧原子与7a位相连。
化合物的noesy谱显示7.19(h-4)与3.15(h-3a),3.07(h-3b)相关,所以进一步说明了五元呋喃环氧原子与7a位而不是3a位相连。
由于化合物9和10消旋体的
表9化合物9和10消旋体的nmr数据归属
化合物11和12消旋体:lupuloneg的结构鉴定数据如下:
无色针状结晶(甲醇),
各个片段的连接方式由hmbc谱来确定,δh3.75(1h,m)与δc209.9(c-1"),160.4(c-7a)相关,则异丙基与羰基碳相连,且异丁酰基连在7位碳上;δh7.28(1h,d,j=8.2hz,h-4)与28.4(c-3)相关,δh3.14-2.99(2h,m)与69.9(c-1'),91.3(c-2),118.5(c-3a)相关,提示五元呋喃环氧原子与7a位相连。
化合物的noesy谱显示7.28(h-4)与3.14-2.99(h-3)相关,所以进一步说明了五元呋喃环氧原子与7a位而不是3a位相连。
由于化合物11和12消旋体的
表10化合物11和12消旋体的nmr数据归属
化合物13和14消旋体:5-deprenyllupulonolc的结构鉴定数据如下:
无色针状结晶(甲醇),
各个片段的连接方式由hmbc谱来确定,δh2.88(1h,dd,j=15.0,6.6hz,h-2"a),2.79(1h,dd,j=15.0,7.3hz,h-2"b)与δc203.5(c-1"),160.7(c-7a)相关,则异丁基与羰基碳相连,且异戊酰基连在7位碳上;δh2.98(2h,d,j=9.1hz,h-3)与69.9(c-1'),90.7(c-2),106.9(c-3a)相关,提示五元呋喃环氧原子与7a位相连。δh3.12(2h,d,j=7.2hz,h-1”')与106.9(c-3a),104.0(c-5),123.3(c-2”')相关,提示异戊烯基与c-4相连,进一步说明了五元呋喃环氧原子与7a位而不是3a位相连。
由于化合物13和14消旋体的
表11化合物13和14消旋体的nmr数据归属
实施例2
(1)啤酒花1000g用95%乙醇加热回流提取3次(用量:10l),减压回收提取液得粗提物;
(2)步骤(1)所得乙醇提取物经硅胶柱色谱法分离,依次以石油醚:丙酮100:1,100:3,100:6,100:9,100:11,100:15,5:1,1:1洗脱,
(3)上述步骤(2)中所得的石油醚:丙酮100:3流分经ods色谱分离,以乙腈/水1:9,2:8,4:6,1:1,6:4为流动相梯度洗脱;
(4)上述步骤(3)中所得乙腈:水(4:6)流分经hplc-rid色谱分离,以乙腈:水(3:7)为流动相洗脱得到新α-酸,化合物9和10消旋体(tr=60min,收率0.011%)和化合物4(tr=75min)(收率0.043%)。化合物9和10消旋体(tr=60min,收率0.011%)和化合物4(tr=75min)(收率0.043%)。化合物9和10消旋体再经手性柱色谱分离,以正己烷:乙醇(90:10)为流动相洗脱得到新的α-酸9(13.642min),10(15.796min)(收率各为0.005%)。
(5)上述步骤(3)中所得乙腈:水(2:8)流分经结晶溶剂乙腈:水(4:6),进行反复重结晶技术得化合物13和14消旋体(收率0.040%)。化合物13和14消旋体再经手性柱色谱分离,以正己烷:乙醇(90:10)为流动相洗脱得到新的α-酸13(10.574min),14(14.078min)(收率各为0.019%)。
(6)上述步骤(2)中所得的石油醚:丙酮100:6流分经ods色谱分离,以乙腈/水1:9,3:7,4:6,1:1,6:4为流动相梯度洗脱;
(7)上述步骤(6)中所得乙腈:水(3:7)流分经hplc-rid色谱分离,以乙腈:水(4:6)为流动相洗脱得到新α-酸,化合物1(tr=48min)(收率0.076%)和化合物5(tr=60min)(收率0.093%)。
(8)上述步骤(2)中所得的石油醚:丙酮100:9流分经ods色谱分离,以乙腈/水1:9,2:8,4:6,1:1,6:4为流动相梯度洗脱;
(9)上述步骤(8)中所得乙腈:水(2:8)流分经hplc-rid色谱分离,以乙腈:水(38:62)为流动相洗脱得到新α-酸,化合物7(tr=75min)(收率0.025%)。
(10)上述步骤(2)中所得的石油醚:丙酮100:11流分经ods色谱分离,以乙腈/水1:9,3:7,1:1,6:4为流动相梯度洗脱;
(11)上述步骤(10)中所得乙腈:水(3:7)流分经hplc-rid色谱分离,以乙腈:水(35:65)为流动相洗脱得到新α-酸,化合物2(tr=50min,收率0.014%),化合物3(tr=64min,收率0.041%),化合物6(tr=40min,收率0.015%),化合物11和12消旋体(tr=70min,收率0.009%)。化合物11和12消旋体再经手性柱色谱分离,以正己烷:乙醇(90:10)为流动相洗脱得到新的α-酸11(13.124min),12(14.964min)(收率各为0.004%)。
(12)上述步骤(2)中所得的石油醚:丙酮5:1流分经ods色谱分离,以乙腈/水1:9,3:7,1:1,6:4为流动相梯度洗脱;
(13)上述步骤(12)中所得乙腈:水(3:7)流分经结晶溶剂乙腈:水(2:8)反复重结晶得新α-酸,化合物8(收率0.043%)。
α-酸类1-14的结构鉴定方法见实施例1。
实施例3
(1)啤酒花2000g用100%乙醇提取2次(用量:16l),减压回收提取液得粗提物;
(2)步骤(1)所得乙醇提取物经硅胶柱色谱法分离,依次以氯仿:丙酮100:1,100:2,100:4,100:6,100:8,100:9,100:11,100:15,5:1洗脱;
(3)上述步骤(2)中所得的氯仿:丙酮100:2流分经ods色谱分离,以乙腈/水1:9,2:8,4:6,1:1,6:4为流动相梯度洗脱;
(4)上述步骤(3)中所得乙腈:水(4:6)流分经hplc-rid色谱分离,以乙腈:水(38:62)为流动相洗脱得到新α-酸,化合物9和10消旋体(tr=42min,收率0.010%)和化合物4(tr=55min)(收率0.044%)。化合物9和10消旋体再经手性柱色谱分离,以正己烷:乙醇(93:7)为流动相洗脱得到新的α-酸9(11.725min),10(13.956min)(收率各为0.005%)。
(5)上述步骤(3)中所得乙腈:水(4:6)流分经结晶溶剂乙腈:水(3:7),进行反复重结晶技术得化合物13和14消旋体(收率0.044%)。化合物13和14消旋体再经手性柱色谱分离,以正己烷:乙醇(93:7)为流动相洗脱得到新的α-酸13(7.972min),14(10.491min)(收率各为0.022%)。
(6)上述步骤(2)中所得的氯仿:丙酮100:4流分经ods色谱分离,以乙腈/水1:9,3:7,1:1,6:4为流动相梯度洗脱;
(7)上述步骤(6)中所得乙腈:水(3:7)流分经hplc-rid色谱分离,以乙腈:水(1:1)为流动相洗脱得到新α-酸,化合物1(tr=35min)(收率0.074%)和化合物5(tr=44min)(收率0.093%)。
(8)上述步骤(2)中所得的氯仿:丙酮100:6流分经ods色谱分离,以乙腈/水1:9,4:6,1:1,6:4为流动相梯度洗脱;
(9)上述步骤(8)中所得乙腈:水(4:6)流分经hplc-rid色谱分离,以乙腈:水(1:1)为流动相洗脱得到新α-酸,化合物7(tr=50min)(收率0.026%)。
(10)上述步骤(2)中所得的氯仿:丙酮100:9流分经ods色谱分离,以乙腈/水2:8,3:7,1:1,6:4为流动相梯度洗脱;
(11)上述步骤(10)中所得乙腈:水(3:7)流分经hplc-rid色谱分离,以乙腈:水(35:65)为流动相洗脱得到新α-酸,化合物2(tr=40min,收率0.012%),化合物3(tr=52min,收率0.035%),化合物6(tr=30min,收率0.014%),化合物11和12消旋体(tr=60min,收率0.012%)。化合物11和12消旋体再经手性柱色谱分离,以正己烷:乙醇(93:7)为流动相洗脱得到新的α-酸11(11.043min),12(12.412min)(收率各为0.006%)。
(12)上述步骤(2)中所得的氯仿:丙酮100:15流分经ods色谱分离,以乙腈/水1:9,2:8,4:6,1:1,6:4为流动相梯度洗脱;
(13)上述步骤(12)中所得乙腈:水(1:9)流分经结晶溶剂乙腈:水(2:8),进行反复重结晶得新α-酸,化合物8(收率0.040%)。
α-酸类1-14的结构鉴定方法见实施例1。
实施例4
(1)啤酒花1500g用80%乙醇提取1次(用量:22.5l),减压回收提取液得粗提物;
(2)步骤(1)所得乙醇提取物经硅胶柱色谱法分离,依次以氯仿:甲醇100:1,100:2.5,100:4,100:6,100:8,100:10,100:12,100:15,5:1洗脱;
(3)上述步骤(2)中所得的氯仿:甲醇100:1流分经ods色谱分离,以甲醇/水3:7,1:1,7:3,9:1为流动相梯度洗脱;
(4)上述步骤(3)中所得甲醇:水(1:1)流分经hplc-rid色谱分离,以甲醇:水(6:4)为流动相洗脱得到新α-酸,化合物9和10消旋体(tr=59min,收率0.011%)和化合物4(tr=74min)(收率0.048%)。化合物9和10消旋体再经手性柱色谱分离,以正己烷:乙醇(92:8)为流动相洗脱得到新的α-酸9(12.739min),10(14.750min)(收率各为0.005%)。
(5)上述步骤(3)中所得甲醇:水(1:1)流分经结晶溶剂甲醇:水(8:2)进行反复重结晶技术得化合物13和14消旋体(收率0.041%)。化合物13和14消旋体再经手性柱色谱分离,以正己烷:乙醇(92:8)为流动相洗脱得到新的α-酸13(9.327min),14(11.576min)(收率各为0.020%)。
(6)上述步骤(2)中所得的氯仿:甲醇100:4流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱;
(7)上述步骤(6)中所得甲醇:水(4:6)流分经hplc-rid色谱分离,以甲醇:水(56:44)为流动相洗脱得到新α-酸类1(tr=62min)(收率0.072%)和5(tr=75min)(收率0.093%)。
(8)上述步骤(2)中所得的氯仿:甲醇100:6流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱;
(9)上述步骤(8)中所得甲醇:水(4:6)流分经hplc-rid色谱分离,以甲醇:水(66:34)为流动相洗脱得到新α-酸类7(tr=90min)(收率0.023%)。
(10)上述步骤(2)中所得的氯仿:甲醇100:6流分经ods色谱分离,以甲醇/水2:8,4:6,1:1,6:4,9:1为流动相梯度洗脱;
(11)上述步骤(10)中所得甲醇:水(1:1)流分经hplc-rid色谱分离,以甲醇:水(1:1)为流动相洗脱得到新α-酸类2(tr=69min,收率0.013%),3(tr=80min,收率0.040%),6(tr=57min,收率0.015%),化合物11和12消旋体(tr=65min,收率0.010%)。化合物11和12消旋体再经手性柱色谱分离,以正己烷:乙醇(92:8)为流动相洗脱得到新的α-酸11(11.872min),12(13.519min)(收率各为0.005%)。
(12)上述步骤(2)中所得的氯仿:甲醇10:1流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱;
(13)上述步骤(12)中所得甲醇:水(4:6)流分经结晶溶剂甲醇:水(6:4)反复重结晶得新α-酸类8(收率0.041%)。
α-酸衍生物1-14的结构鉴定方法见实施例1。
实施例5
(1)啤酒花2000g用75%乙醇提取1次(用量:10l),减压回收提取液得粗提物;
(2)步骤(1)所得乙醇提取物经硅胶柱色谱法分离,依次以二氯甲烷:丙酮100:1,100:2,100:3,100:4,100:6,100:8,100:9,100:11,100:15,5:1洗脱;
(3)上述步骤(2)中所得的二氯甲烷:丙酮100:2流分经ods色谱分离,以甲醇/水3:7,1:1,7:3为流动相梯度洗脱;
(4)上述步骤(3)中所得甲醇:水(1:1)流分经hplc-rid色谱分离,以甲醇:水(67:33)为流动相洗脱得到新α-酸,化合物9和10消旋体(tr=50min,收率0.011%)和化合物4(tr=68min)(收率0.045%)。化合物9和10消旋体再经手性柱色谱分离,以正己烷:乙醇(94:6)为流动相洗脱得到新的α-酸9(10.965min),10(13.226min)(收率各为0.005%)。
(5)上述步骤(3)中所得甲醇:水(7:3)流分经结晶溶剂甲醇:水(9:1),进行反复重结晶技术得化合物13和14消旋体(收率0.041%)。化合物13和14消旋体再经手性柱色谱分离,以正己烷:乙醇(94:6)为流动相洗脱得到新的α-酸13(7.720min),14(9.997min)(收率各为0.019%)。
(6)上述步骤(2)中所得的二氯甲烷:丙酮100:3流分经ods色谱分离,以甲醇/水4:6,1:1,6:4为流动相梯度洗脱;
(7)上述步骤(6)中所得甲醇:水(4:6)流分经hplc-rid色谱分离,以甲醇:水(6:4)为流动相洗脱得到新α-酸,化合物1(tr=61min)(收率0.073%)和化合物5(tr=78min)(收率0.093%)。
(8)上述步骤(2)中所得的二氯甲烷:丙酮100:4流分经ods色谱分离,以甲醇/水2:8,4:6,1:1,6:4为流动相梯度洗脱;
(9)上述步骤(8)中所得甲醇:水(1:1)流分经hplc-rid色谱分离,以甲醇:水(77:23)为流动相洗脱得到新α-酸,化合物7(tr=69min)(收率0.025%)。
(10)上述步骤(2)中所得的二氯甲烷:丙酮100:8流分经ods色谱分离,以甲醇/水3:7,1:1,7:3为流动相梯度洗脱;
(11)上述步骤(10)中所得甲醇:水(7:3)流分经hplc-rid色谱分离,以甲醇:水(6:4)为流动相洗脱得到新α-酸,化合物2(tr=65min,收率0.013%),化合物3(tr=76min,收率0.035%),化合物6(tr=48min,收率0.014%),化合物11和12消旋体(tr=80min,收率0.009%)。化合物11和12消旋体再经手性柱色谱分离,以正己烷:乙醇(94:6)为流动相洗脱得到新的α-酸11(10.558min),12(11.804min)(收率各为0.004%)。
(12)上述步骤(2)中所得的二氯:丙酮100:15流分经ods色谱分离,以甲醇/水2:8,4:6,1:1,6:4,9:1为流动相梯度洗脱;
(13)上述步骤(12)中所得甲醇:水(1:1)流分经结晶溶剂甲醇:水(6:4)反复重结晶技术得新α-酸,化合物8(收率0.042%)。
α-酸类1-14的结构鉴定方法见实施例1。
实施例6
(1)啤酒花1500g用95%乙醇提取1次(用量:30l),减压回收提取液得粗提物;
(2)步骤(1)所得乙醇提取物经硅胶柱色谱法分离,依次以二氯甲烷:甲醇100:1,100:3,100:5,100:6,100:9,100:12,100:15,5:1洗脱;
(3)上述步骤(2)中所得的二氯甲烷:甲醇100:1流分经ods色谱分离,以乙腈/水1:9,3:7,1:1,7:3为流动相梯度洗脱;
(4)上述步骤(3)中所得乙腈:水(3:7)流分经hplc-rid色谱分离,以乙腈:水(1:1)为流动相洗脱得到新α-酸,化合物9和10消旋体(tr=40min,收率0.013%)和化合物4(tr=55min)(收率0.047%)。化合物9和10消旋体再经手性柱色谱分离,以正己烷:乙醇(93:7)为流动相洗脱得到新的α-酸9(11.865min),10(13.933min)(收率各为0.006%)。
(5)上述步骤(3)中所得乙腈:水(1:1)流分经结晶溶剂乙腈:水(4:6),进行反复重结晶技术得化合物13和14消旋体(收率0.039%)。化合物13和14消旋体再经手性柱色谱分离,以正己烷:乙醇(93:7)为流动相洗脱得到新的α-酸13(8.150min),14(10.608min)(收率各为0.018%)。
(6)上述步骤(2)中所得的二氯甲烷:甲醇100:3流分经ods色谱分离,以乙腈/水1:9,2:8,4:6,1:1,6:4为流动相梯度洗脱;
(7)上述步骤(6)中所得乙腈:水(2:8)流分经hplc-rid色谱分离,以乙腈:水(33:67)为流动相洗脱得到新α-酸类1(tr=54min)(收率0.073%)和5(tr=62min)(收率0.095%)。
(8)上述步骤(2)中所得的二氯甲烷:甲醇100:5流分经ods色谱分离,以乙腈/水1:9,2:8,1:1,7:3为流动相梯度洗脱;
(9)上述步骤(8)中所得乙腈:水(2:8)流分经hplc-rid色谱分离,以乙腈:水(38:62)为流动相洗脱得到新α-酸类7(tr=68min)(收率0.025%)。
(10)上述步骤(2)中所得的二氯甲烷:甲醇100:6流分经ods色谱分离,以乙腈/水1:9,3:7,1:1,6:4为流动相梯度洗脱;
(11)上述步骤(10)中所得乙腈:水(3:7)流分经hplc-rid色谱分离,以乙腈:水(4:6)为流动相洗脱得到新α-酸类2(tr=42min,收率0.013%),3(tr=54min,收率0.041%),6(tr=30min,收率0.016%),化合物11和12消旋体(tr=65min,收率0.013%)。化合物11和12消旋体再经手性柱色谱分离,以正己烷:乙醇(93:7)为流动相洗脱得到新的α-酸11(11.045min),12(12.610min)(收率各为0.006%)。
(12)上述步骤(2)中所得的二氯甲烷:甲醇10:1流分经ods色谱分离,以乙腈/水1:9,2:8,4:6,1:1,6:4为流动相梯度洗脱;
(13)上述步骤(12)中所得乙腈:水(2:8)流分经结晶溶剂乙腈:水(3:7)反复重结晶得新α-酸类8(收率0.043%)。
α-酸衍生物1-14的结构鉴定方法见实施例1。
实施例7实施例1-6中制备得到的新α-酸类衍生物1-14的抗神经炎症活性测试
(1)实验原理:小胶质细胞活化介导的慢性炎症反应是神经退行性疾病的发生、发展过程中的重要环节,抑制小胶质细胞的激活可能成为药物发现的一个新的靶点。lps激活小胶质细胞释放no、促炎症细胞因子和活性氧等。本实验通过建立体外lps激活bv2小胶质细胞异常活化的筛选模型,以激活小胶质细胞释放no为指标,评价新α-酸类衍生物1-14的抗炎活性。
(2)实验方法:
①小鼠小胶质细胞系bv2的培养
细胞培养和模型建立中使用的所有玻璃器皿及金属器械(培养瓶,移液管,溶液瓶等),均经过121℃高压灭菌30min,以彻底去除污染的lps。以dmem培养基作为基础配制成内含10%胎牛血清及50μm2-巯基乙醇的细胞培养液。小胶质细胞以约4×105cells/ml的浓度在5%co2,37℃培养瓶中传代培养,至第三天贴壁细胞约占培养瓶底面积50-60%,以胰酶消化贴壁细胞,传代至另一培养瓶。以-80℃超低温冰箱冻存复苏后的bv2作为第一代,选择第3-8代bv2细胞进行实验。
②药物配制方法
测试化合物均为粉末状,用dmso溶解。配成母液,浓度为50mm,储存于-20℃。临用时用dmem培养液将其进行稀释,依次稀释为100μm、30μm、10μm、3μm、1μm。dmso终浓度<1‰。
③griess法检测化合物对lps激活小胶质细胞的抑制作用
取对数生长期的bv2小胶质细胞,用含5%胎牛血清的新鲜dmem培养基将细胞密度调至3×105cells/ml,接种于96孔板内,100μl/well,于37℃,5%co2的培养箱内培养。细胞贴壁培养24h后换成无血清的新鲜培养液,同时进行加药处理。每种化合物设剂量1、3、10、30、100μm与lps共同作用。同时设空白对照。各给药组中lps终浓度为100ng/ml。细胞加药后继续培养24h后,收集上清液,griess比色法检测上清液中no2-含量。
④mtt法检测化合物对小胶质细胞细胞成活率的影响
取对数生长期培养的bv2小胶质细胞,用含5%胎牛血清的新鲜dmem培养基将细胞密度调至3×105cells/ml,接种于96孔板内,100μl/well,于37℃,5%co2的培养箱内培养。细胞贴壁培养24h后换成新鲜培养液,同时进行加药处理。每种化合物设剂量1、3、10、30、100μm与lps共同作用。同时设空白对照。各给药组中lps终浓度为100ng/ml。细胞加药后继续培养24h,然后向细胞液中加入mtt溶液,10μl/well,将细胞与0.25mg/mlmtt于37℃下共同孵育3h,吸除培养液,然后加入100μl的dmso溶液,测定其光密度od值。数据处理,利用酶标仪相应软件进行数据处理,计算每一种样品6个孔od值的平均值,利用平均值按如下公式计算细胞成活率(cellviability,cv%)。
细胞成活率%=样品组od值的平均值/空白对照组od值的平均值×100%
⑤统计方法
全部资料采用spss(13.0)统计软件包进行检验分析。结果用平均值±标准误表示,评价整体性差异,组间均数采用one-wayanova分析法进行方差齐性分析,并结合dunnett’stest分析方法进行组间比较。多样本方差齐性检验采用levene检验,当p>0.05,方差是齐的,采用dunnett’s双侧t检验多组间均数的差异,当p<0.05,方差不齐,采用dunnettt3检验多组间均数的差异。
⑥ic50的计算方法
将各剂量和抑制率等参数用非线性回归拟合计算ic50
(3)实验结果:见表12
表12新α-酸类衍生物1-14抑制小胶质细胞活化作用实验结果
significance:*p<0.05,**p<0.01,***p<0.001与lps诱导组相比;###p<0.001与对照组相比.
结果可知,实施例1-6中制备得到的新α-酸类化合物1,9,10,11,12,13,14(30μm,100μm);3,5,7(100μm)能够显著的抑制lps诱导的过度活化的bv2小胶质细胞no的释放;化合物2,4,6,8随着浓度的增加,表现出了潜在的抑制no的释放的活性。
实施例13实施例1-6中制备得到的新α-酸类衍生物1-14保护双氧水诱导的hepg2肝细胞损伤的作用
(1)实验原理:体外肝细胞损伤模型的建立是对药物保肝作用筛选及分子作用机制研究所必须的。其中,肝细胞氧化损伤模型有ccl4和h2o2,由于ccl4诱导模型作用更持久,细胞毒性更大,实际操作可控性差,损伤效果不稳定,损伤也不均匀,而h2o2诱导模型作用更直接,以短时间急性损伤为主,故本实验选h2o2作为作为损伤的模型。h2o2是一种强氧化剂,是经典的诱导细胞发生氧化损伤的试剂,也是体内最常见的一种活性氧,在多种组织损伤中发挥了关键性作用。h2o2作用肝细胞后,它能被细胞色素p-450代谢分解,生成·oh自由基,进而攻击细胞生物膜中的不饱和脂肪酸,引发脂质过氧化反应,大量消耗细胞内源性还原gsh,增加过氧化物mda的积累,最终导致细胞死亡。目前,建立肝细胞损伤体外模型细胞以l-02(人的肝正常细胞)和hepg2两种多见。本实验中应用hepg2细胞系统评价14个新的衍生物保护肝细胞的作用。
(2)实验方法
①主要试剂的配制
维生素e储备液的配制:精密20mg,加入232.2μl的dmso溶解配成200mm的储备液,-20℃保存备用。
过氧化氢储备液的配制:精密吸取3.3μl的30%h2o2,然后用dmem定容到10μl,制备成3200μm的储备液,-20℃保存备用。
②实验分组:正常对照组,h2o2模型组,维生素e+h2o2,化合物(1,10,50μm)+h2o2,共计6组,每组均设3个复孔。
③实验步骤:
取对数生长期hepg2细胞,以6×104个/ml接种于96孔培养板中,于37℃、5%co2培养箱中培养24h;以维生素e(500μm)和化合物(1,10,50μm)预处理细胞6h;加入h2o2(200μm)继续培养2h;用mtt法在490nm波长处测细胞存活力。
存活率(%)=(实验组od-空白组od)/(对照组od-空白组od)×100%
保护率(%)=(样品组od-损伤组od)/(正常组od-损伤组od)×100%
④统计方法
用spss17.0统计软件,各组数据用mean±se表示,用单因素方差进行分析(anova),采用lsd进行组间均数的两两比较,p<0.05为差异具有统计学意义。
(4)实验结果
如图1,图2中所示
结果显示,实施例1-6中制备得到的新α-酸类衍生物2,8,9,10,11,12,13,14能够显著的保护h2o2引起的hepg2肝细胞氧化损伤;化合物7具有中等强度的肝细胞保护活性;化合物1,3,4,5,6在测试浓度下表现出一定程度的肝保护活性。
实施例14实施例1-6中制备得到的新α-酸类衍生物1-14保护双氧水诱导的l02肝细胞损伤的作用
(1)实验原理:如上所述。本实验中应用l02细胞系统评价14个新的化合物保护肝细胞的作用。
(2)实验方法
①主要试剂的配制
维生素e储备液的配制:精密20mg,加入232.2μl的dmso溶解配成200mm的储备液,-20℃保存备用。
过氧化氢储备液的配制:精密吸取3.3μl的30%h2o2,然后用dmem定容到10μl,制备成3200μm的储备液,-20℃保存备用。
②实验分组:正常对照组,h2o2模型组,维生素e+h2o2,化合物(1,10,50μm)+h2o2,共计6组,每组均设3个复孔。
③实验步骤:
取对数生长期l-02细胞,以8×104个/ml密度接种于96孔培养板中,于37℃、5%co2培养箱中培养24h;以维生素e(500μm)和化合物(1,10,50μm)预处理细胞6h;加入h2o2(100μm)继续培养2h;用mtt法在490nm波长处测细胞存活力。
存活率(%)=(实验组od-空白组od)/(对照组od-空白组od)×100%
保护率(%)=(样品组od-损伤组od)/(正常组od-损伤组od)×100%
④统计方法
用spss17.0统计软件,各组数据用mean±se表示,用单因素方差进行分析(anova),采用lsd进行组间均数的两两比较,p<0.05为差异具有统计学意义。
(5)实验结果
如图3,图4中所示
结果显示,实施例1-6中制备得到的新α-酸类化合物2,8,9,10,11,12,13,14能够显著的保护h2o2引起的l-02肝细胞氧化损伤;化合物7具有中等强度的肝细胞保护活性;化合物3,4,5,6在测试浓度下表现出一定程度的肝保护活性。
- 还没有人留言评论。精彩留言会获得点赞!