一种细胞糖转运通道抑制剂
本发明属于生物医药技术领域,涉及一种抑制细胞的糖营养吸收,靶向肿瘤沃伯格效应的一种细胞糖转运通道抑制剂,其中细胞糖转运通道简称glut。
背景技术:
目前,恶性肿瘤是威胁人类生命健康的一类重大疾病,对付肿瘤最好的策略就是通过靶向肿瘤特异性的生物靶点,在不伤害正常细胞和组织的前提下抑制肿瘤生长。
靶向抗肿瘤的方法有很多,其基本原理是利用肿瘤细胞或肿瘤组织特异性存在,或者与正常细胞或正常组织具有显著差异的肿瘤生物标志物,通过选择性识别、抑制这些生物标志物而达到治疗肿瘤的目的。研究表明,恶性肿瘤之所以能够持续增殖、扩散和转移,其重要的生理机制之一是不断吸收和摄取它所需要的特殊营养。这些营养包括,糖分子、氨基酸、维生素、核苷等。德国生物化学家奥托·沃伯格发现健康细胞能有效利用有氧代谢,从有限的糖类营养源制备足够的能量,而大多数肿瘤细胞即便在氧气充足的情况下也依然依靠大量的糖吸收和糖代谢为自身提供能源,这种现象被称为沃伯格效应。沃伯格因此于1931年获得了生理学或医学诺贝尔奖(参考文献:warburg,o.science,123:309(1956);"thenobelprizeinphysiologyormedicine1931".nobelprize.org.<http://www.nobelprize.org/nobel_prizes/medicine/laureates/1931/>)。
基于肿瘤沃伯格效应,由于肿瘤细胞与健康细胞相比特异性高表达糖转运通道glut,因此以glut为肿瘤特异性标志物有望实现对肿瘤的靶向治疗。如式(a)所示,作为现有技术,文献报道了化合物1作为glut抑制剂能够阻止血癌细胞hl-60和u-937的糖吸收和糖积蓄(salasm.,etal.,am.j.physiol.cellphysiol.,305:c90(2013))。另有文献报道化合物2能够抑制抑制卵巢癌的糖酵解代谢和组织肿瘤细胞生长(yibaoma,etal.,cancers,11(1):33(2019))。另有报道,化合物3作为glut选择性抑制剂,但没有验证其在抑制肿瘤细胞中的作用(siebeneicherh,etal.chemmedchem.2016,11(20):2261-2271)。
这些通过不同药物筛选方法获得的glut抑制剂为进一步验证这些先导化合物的临床应用提供了物质基础。但还需要更多有验证的具有glut选择性的抑制剂的开发。
技术实现要素:
本发明的目的是提供一种能够有效抑制细胞糖转运通道glut,利用肿瘤沃伯格效应,即肿瘤细胞与正常细胞相比过度表达glut的特征,实现抑制肿瘤生长的目的的细胞糖转运通道抑制剂。
本发明的第二个目的是提供一种细胞糖转运通道抑制剂在制备抗肿瘤药物中用途或制备抑制细胞糖转运通道glut的试剂的用途。
本发明的第三个目的是提供一种抑制肿瘤的药物组合物。
本发明的第四个目的是提供一种抑制肿瘤的药物组合物在制备抗肿瘤药物中用途。
本发明的技术方案概述如下:
一种细胞糖转运通道抑制剂,是结构如式i所示:
其中,
xr1和xr2相同或不同;
xr1表示苯环不同位置的单取代或双取代;
xr2表示苯环不同位置的单取代或双取代;
x选自o,s或nh;
r1为氢原子,c1-c7直链烷基或c3-c8支链烷基;
r2为氢原子,c1-c7直链烷基或c3-c8支链烷基。
y选自卤素,芳基磺酰基,芳基亚磺酰基,芳巯基或胺基磺酰基。
优选地,xr1和xr2相同;
xr1表示苯环不同位置的单取代或双取代;
xr2表示苯环不同位置的单取代或双取代;
x为o;
r1为氢原子;
r2为氢原子;
y为f,cl,phso2,phso,phs或so2n(me)2。
上述一种细胞糖转运通道抑制剂在制备抗肿瘤药物中用途或制备抑制细胞糖转运通道glut的试剂的用途。
一种抑制肿瘤的药物组合物,包括上述一种细胞糖转运通道抑制剂或其药学上可接受的盐或其溶剂化物。
上述一种抑制肿瘤的药物组合物在制备抗肿瘤药物中用途。
本发明具有以下的有益效果:
实验证明,本发明的一种细胞糖转运通道抑制剂能靶向细胞糖转运通道,通过直接阻断glut通道和肿瘤对糖营养成分的摄取而抑制肿瘤细胞增殖。
附图说明
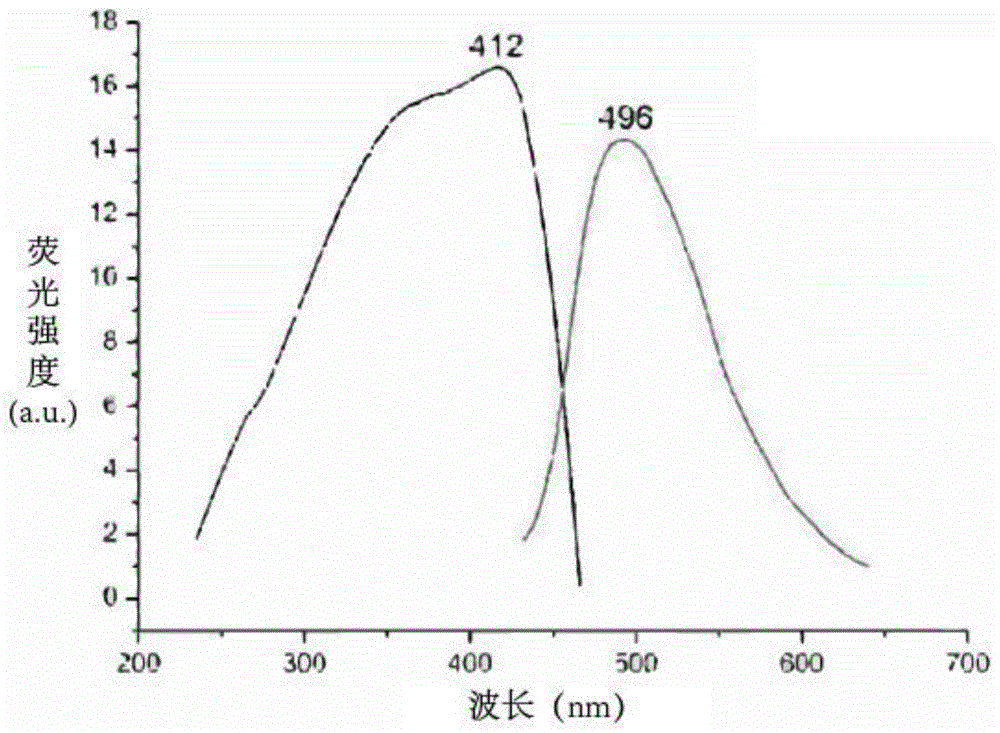
图1为一种细胞糖转运通道抑制剂(i)-(a-1)-1的荧光吸收图谱;
图2为一种细胞糖转运通道抑制剂(i)-(a-2)-1的荧光吸收图谱;
图3为一种细胞糖转运通道抑制剂(i)-(a-3)-1的荧光吸收图谱;
图4为人肺癌a549及乳腺癌mda-mb-468细胞表达glut的测试结果。
具体实施方式
下面通过具体实施例对本发明作进一步详细的说明,是对本发明的解释而不是限定。
一种细胞糖转运通道抑制剂,以下简称glut抑制剂。
实施例1
glut抑制剂(i)-(a-1)-1的合成:
1、4-氯-6-硝基-7-乙酰氨基苯并-2-氧杂-1,3-二唑(ⅱ)的合成,其合成路线如下:
4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑(xiv,678.3mg,3.0mmol)溶解于12ml乙酸酐中。在0℃搅拌下,向混合物中缓慢滴加4ml浓硝酸。并在0℃下继续搅拌3小时后,加入冰水。搅拌10min后过滤,滤饼用冷水洗涤3次,得到黄色固体(ⅱ)753.3mg,产率59%。1hnmr(600mhz,dmso-d6):δ11.51(s,1h),8.19(s,1h),2.19(s,3h).
反应式为:
2、4-氯-6-硝基-7-氨基苯并-2-氧杂-1,3-二唑(ⅲ)的合成,其合成路线如下:
将ⅱ(256.6mg,1.0mmol)加入50%wt.h2so4水溶液(3ml),混合物在100℃下搅拌2小时。将混合物冷却至室温,用饱和碳酸氢钠水溶液调ph至8,乙酸乙酯萃取(20ml×2),有机相用无水硫酸钠干燥。用旋转蒸发仪除去溶剂,得到黄色固体(ⅲ)191.1mg,收率89%。1hnmr(600mhz,dmso-d6):δ9.83(s,1h),9.35(s,1h),8.14(s,1h).
反应式为:
3、4-氯-6,7-二氨基苯并-2-氧杂-1,3-二唑(ⅳ)的合成,其合成路线如下:
在ⅲ(107.3mg,0.5mmol)的thf(10ml)溶液中加入10%pd/c(20mg)。将混合物置于氢气中在25℃下搅拌12小时。混合物用硅藻土过滤,thf洗涤2次,用旋转蒸发仪将溶剂除去,经硅胶柱色谱纯化(石油醚∶乙酸乙酯=4∶1至2∶1),得到红色固体(ⅳ)64.6mg,收率70%。
1hnmr(400mhz,dmso-d6):δ7.29(s,1h),5.38(s,2h),5.16(s,2h)
反应式为:
4、4-叔丁基二甲基硅氧基苯甲酰氯(ⅶ)的合成:
将4-羟基苯甲酸(ⅴ,3.0g,21.7mmol),叔丁基二甲基氯硅烷(tbscl)(7.5g,50.0mmol)和咪唑(3.4g,50.0mmol)溶于无水n,n-二甲基甲酰胺(dmf)(30ml)中,在室温下搅拌24h。反应完成后,将丙酮(150ml)加入到反应混合物中,过滤,收集滤液,减压浓缩,所得残余物溶于乙酸乙酯(50ml)中,饱和氯化铵水溶液洗涤3次,无水硫酸钠干燥,减压除去溶剂,得到无色液体。
将所得无色液体溶于冰醋酸(50ml),四氢呋喃(15ml)和水(15ml)的混合溶剂中,在室温下搅拌4h。加入乙酸乙酯(60ml)和水(60ml),用饱和碳酸氢钠水溶液洗涤,收集有机相,用无水硫酸钠干燥。用旋转蒸发仪将溶剂蒸干,所得残余物在高真空下干燥,得到白色固体4-叔丁基二甲基硅氧基苯甲酸(ⅵ)3.7g,产率68%。
在氮气的保护下加入ⅵ(2.5g,10.0mmol)),n,n-二甲基甲酰胺(dmf)(2滴)和无水二氯甲烷(dcm)(20ml)。在0℃的条件下,逐滴向反应液中加入草酰氯(1.0ml,12.0mmol),在室温下搅拌3h,用旋转蒸发仪将溶剂蒸干,所得ⅶ无需进一步纯化立即用于下一步反应。
反应式为:
5、(i)-(a-1)-1的合成
在ⅳ(184.6mg,0.6mmol)的四氢呋喃(4ml)溶液中依次加入ⅶ(650.1mg,2.4mmol)的四氢呋喃(4ml)溶液、碳酸钾(207.3mg,1.5mmol),将反应液置于封管中在80℃下搅拌12小时。反应液冷却至室温后过滤,用dcm洗滤渣2次,用旋转蒸发仪将反应液的溶剂除去,经硅胶柱色谱纯化(石油醚∶乙酸乙酯=40∶1),得到淡黄色固体。将固体溶于thf(4ml)中,室温搅拌下滴加适量1m四正丁基氟化铵(tbaf)的四氢呋喃(thf)溶液,搅拌0.5h后,加入二氯甲烷(20ml),依次用水,饱和nacl溶液洗涤,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱色谱纯化(石油醚∶乙酸乙酯=1∶1),得到淡黄色固体(i)-(a-1)-153.4mg,收率21%。
反应式为:
实施例2
glut抑制剂(i)-(a-2)-1的合成:
1、3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ)的合成:
将3-羟基苯甲酸(ⅷ,3.0g,21.7mmol),叔丁基二甲基氯硅烷(tbscl)(7.5g,50.0mmol)和咪唑(3.4g,50.0mmol)溶于无水n,n-二甲基甲酰胺(dmf)(30ml)中,在室温下搅拌24h。反应完成后,将丙酮(150ml)加入到反应混合物中,过滤,收集滤液,减压浓缩,所得残余物溶于乙酸乙酯(50ml)中,饱和氯化铵水溶液洗涤3次,无水硫酸钠干燥,减压除去溶剂,得到无色液体。
将所得无色液体溶于冰醋酸(50ml),四氢呋喃(15ml)和水(15ml)的混合溶剂中,在室温下搅拌4h。加入乙酸乙酯(60ml)和水(60ml),用饱和碳酸氢钠水溶液洗涤,收集有机相,用无水硫酸钠干燥。用旋转蒸发仪将溶剂蒸干,所得残余物在高真空下干燥,得到白色固体3-叔丁基二甲基硅氧基苯甲酸(ⅸ)3.7g,产率87%。
在氮气的保护下加入ⅸ(2.5g,10.0mmol)),n,n-二甲基甲酰胺(dmf)(2滴)和无水二氯甲烷(dcm)(20ml)。在0℃的条件下,逐滴向反应液中加入草酰氯(1.0ml,12.0mmol),在室温下搅拌3h,用旋转蒸发仪将溶剂蒸干,所得3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ)无需进一步纯化立即用于下一步反应。
反应式为:
2、(i)-(a-2)-1的合成
在ⅳ(184.6mg,0.6mmol)的四氢呋喃(4ml)溶液中依次加入ⅹ(650.1mg,2.4mmol)的四氢呋喃(4ml)溶液、碳酸钾(207.3mg,1.5mmol),将反应液置于封管中在80℃下搅拌12小时。反应液冷却至室温后过滤,用dcm洗滤渣2次,用旋转蒸发仪将反应液的溶剂除去,经硅胶柱色谱纯化(石油醚∶乙酸乙酯=40∶1),得到淡黄色固体。将固体溶于thf(4ml)中,室温搅拌下滴加适量1m四正丁基氟化铵(tbaf)的四氢呋喃(thf)溶液,搅拌0.5h后,加入二氯甲烷(20ml),依次用水,饱和nacl溶液洗涤,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱色谱纯化(石油醚∶乙酸乙酯=1∶1),得到淡黄色固体(i)-(a-2)-153.4mg,收率30%。
反应式为:
实施例3
glut抑制剂(i)-(a-3)-1的合成:
1、3,5-二(叔丁基二甲基硅氧基)苯甲酰氯(xiii)的合成:
将3,5-二羟基苯甲酸(ⅺ,3.1g,20.1mmol),叔丁基二甲基氯硅烷(tbscl)(10.6g,70.4mmol)和咪唑(4.8g,70.4mmol)溶于无水n,n-二甲基甲酰胺(dmf)(30ml)中,在室温下搅拌24h。反应完成后,将丙酮(150ml)加入到反应混合物中,过滤,收集滤液,减压浓缩,所得残余物溶于乙酸乙酯(50ml)中,饱和氯化铵水溶液洗涤3次,无水硫酸钠干燥,减压除去溶剂,得到无色液体。
将所得无色液体溶于冰醋酸(50ml),四氢呋喃(15ml)和水(15ml)的混合溶剂中,在室温下搅拌4h。加入乙酸乙酯(60ml)和水(60ml),用饱和碳酸氢钠水溶液洗涤,收集有机相,用无水硫酸钠干燥。用旋转蒸发仪将溶剂蒸干,所得残余物在高真空下干燥,得到白色固体3,5-二(叔丁基二甲基硅氧基)苯甲酸(ⅻ)4.0g,产率52%。
在氮气的保护下加入3,5-二(叔丁基二甲基硅氧基)苯甲酸(ⅻ,3.8g,10.0mmol)),n,n-二甲基甲酰胺(dmf)(2滴)和无水二氯甲烷(dcm)(20ml)。在0℃的条件下,逐滴向反应液中加入草酰氯(1.0ml,12.0mmol),在室温下搅拌3h,用旋转蒸发仪将溶剂蒸干,所得3-叔丁基二甲基硅氧基苯甲酰氯(xiii)无需进一步纯化立即用于下一步反应。
反应式为:
2、(i)-(a-3)-1的合成
在4-氯-6,7-二氨基苯并-2-氧杂-1,3-二唑(ⅳ,184.6mg,0.6mmol)的四氢呋喃(4ml)溶液中依次加入3-叔丁基二甲基硅氧基苯甲酰氯(ⅹⅲ,918.4mg,2.4mmol)的四氢呋喃(4ml)溶液、碳酸钾(207.3mg,1.5mmol),将反应液置于封管中在80℃下搅拌12小时。反应液冷却至室温后过滤,用dcm洗滤渣2次,用旋转蒸发仪将反应液的溶剂除去,经硅胶柱色谱纯化(石油醚∶乙酸乙酯=100∶1),得到淡黄色固体。将固体溶于thf(4ml)中,室温搅拌下滴加适量1m四正丁基氟化铵(tbaf)的四氢呋喃(thf)溶液,搅拌0.5h后,加入二氯甲烷(20ml),依次用水,饱和nacl溶液洗涤,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱色谱纯化(乙酸乙酯∶甲醇=20∶1),得到淡黄色固体(i)-(a-3)-179.4mg,收率29%。
反应式为:
实施例4
glut抑制剂(i)-(a-1)-2的合成遵循实施例1的方法合成,将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换为4-氟-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它同实施例1,制备出glut抑制剂(i)-(a-1)-2。
实施例5
glut抑制剂(i)-(a-2)-2的合成:
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-氟-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-氟-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-氟-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例2制备的3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ),按实施例2的步骤反应,制备出glut抑制剂(i)-(a-2)-2。
实施例6
glut抑制剂(i)-(a-3)-2的合成:
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-氟-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-氟-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-氟-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例3制备的3,5-二(叔丁基二甲基硅氧基)苯甲酰氯(xiii),按实施例3的步骤反应,制备出glut抑制剂(i)-(a-3)-2。
实施例7
glut抑制剂(i)-(a-1)-3的合成遵循实施例1的方法合成,将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换为4-苯磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它同实施例1,制备出glut抑制剂(i)-(a-1)-3。
实施例8
glut抑制剂(i)-(a-2)-3的合成:
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-苯磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-苯磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-苯磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例2制备的3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ),按实施例2的步骤反应,制备出glut抑制剂(i)-(a-2)-3。
实施例9
glut抑制剂(i)-(a-3)-3的合成
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-苯磺酰基--7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-苯磺酰基--6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-苯磺酰基--6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例3制备的3,5-二(叔丁基二甲基硅氧基)苯甲酰氯(xiii),按实施例3的步骤反应,制备出glut抑制剂(i)-(a-3)-3。
实施例10
glut抑制剂(i)-(a-1)-4的合成遵循实施例1的方法合成,将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换为4-苯亚磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它同实施例1,制备出glut抑制剂(i)-(a-1)-4。
实施例11
glut抑制剂(i)-(a-2)-4的合成:
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-苯亚磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-苯亚磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-苯亚磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例2制备的3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ),按实施例2的步骤反应,制备出glut抑制剂(i)-(a-2)-4。
实施例12
glut抑制剂(i)-(a-3)-4的合成
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-苯亚磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-苯亚磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-苯亚磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例3制备的3,5-二(叔丁基二甲基硅氧基)苯甲酰氯(xiii),按实施例3的步骤反应,制备出glut抑制剂(i)-(a-3)-4。
实施例13
glut抑制剂(i)-(a-1)-5的合成
遵循实施例1的方法合成,将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换为4-苯巯基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它同实施例1,制备出glut抑制剂(i)-(a-1)-5。
实施例14
glut抑制剂(i)-(a-2)-5的合成:
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-苯巯基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-苯巯基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-苯巯基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例2制备的3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ),按实施例2的步骤反应,制备出glut抑制剂(i)-(a-2)-5。
实施例15
glut抑制剂(i)-(a-3)-5的合成
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-苯巯基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-苯巯基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-苯巯基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例3制备的3,5-二(叔丁基二甲基硅氧基)苯甲酰氯(xiii),按实施例3的步骤反应,制备出glut抑制剂(i)-(a-3)-5。
实施例16
glut抑制剂(i)-(a-1)-6的合成遵循实施例1的方法合成,将实施例1的
4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换为4-二甲基氨基磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它同实施例1,制备出glut抑制剂(i)-(a-1)-6。
实施例17
glut抑制剂(i)-(a-2)-6的合成:
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-二甲基氨基磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-二甲基氨基磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-二甲基氨基磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例2制备的3-叔丁基二甲基硅氧基苯甲酰氯(ⅹ),按实施例2的步骤反应,制备出glut抑制剂(i)-(a-2)-6。
实施例18
glut抑制剂(i)-(a-3)-6的合成
将实施例1的4-氯-7-乙酰氨基苯并-2-氧杂-1,3-二唑替换成4-二甲基氨基磺酰基-7-乙酰氨基苯并-2-氧杂-1,3-二唑,其它步骤同实施例1,制备出4-二甲基氨基磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑;再将4-二甲基氨基磺酰基-6,7-二氨基苯并-2-氧杂-1,3-二唑与实施例3制备的3,5-二(叔丁基二甲基硅氧基)苯甲酰氯(xiii),按实施例3的步骤反应,制备出glut抑制剂(i)-(a-3)-6。
表1列出了部分通式ⅰ化合物的结构
表1
表2列出了表1各化合物的核磁共振、质谱及荧光光谱数据
表2
试验例1:本发明代表化合物的其他光谱测试
1)实验仪器:thermoscientificvarioskanlux。
2)实验化合物:以上各实施例制备的glut抑制剂化合物。
测试溶剂:dmso:pbs(ph=7.2)=1:9(v:v)
3)实验步骤
(1)称取各测试化合物。
(2)用dmso溶解化合物至10mm,涡旋1min,使化合物充分溶解。
(3)用pbs-dmso(1:1,体积比)稀释化合物至500um,涡旋溶解。取不同化合物与测试溶剂各100ul,加入到黑色底透96孔培养板中,用多功能酶标仪检测吸收光谱、激发光谱和发射光谱。
4)数据处理:用originpro8.5处理,导出吸收谱、发射谱、和吸收-发射合谱。
5)结果:如表2所示。代表性glut抑制剂的激发和发射光谱谱图如附图(图1至图3)所示。
试验例2:glut抑制剂对glut1的抑制作用
依据文献报导人和哺乳动物的红细胞主要表达glut1糖转运通道蛋白进行糖吸收(m.mueckler,c.caruso,s.a.baldwin,i.blench,h.r.morris,w.j.allard,g.e.lienhard,h.f.lodish,science,1985,229,941;(b)montel-hagen,s.kinet,n.manel,c.mongellaz,r.prohaska,j.l.battini,j.delaunay,m.sitbon,n.taylor,cell,2008,132,1039),本实验采用人血红细胞和2-脱氧葡萄糖细胞吸收测试试剂盒(cosmobioco.ltd.,日本)针对本发明glut抑制剂分子进行glut抑制作用评价。具体实验方法和结果如下:
人红细胞采用常规方法采集:枸橼酸抗凝采集的肘静脉全血10ml,于1500r/min离心5分钟,弃去血浆和红细胞表面的白细胞,用10ml磷酸盐缓冲液(ph7.4)洗涤细胞3次(1500r/min,4分钟),红细胞于磷酸盐缓冲液中保存于4度冰箱中待用。将上述红细胞缓冲液悬液计数后以5x106每孔加于96孔板中,设置:①空白对照组每孔加入溶于pbs(ph7.2)中的2-脱氧葡萄糖(购于sigma)200微升(终浓度为1mm);②阳性对照组添加含有终浓度为1mm的2-脱氧葡萄糖和10μm细胞松弛素b(购于sigma)(总体积200微升);③测试组:分别添加含有终浓度1mm的2-脱氧葡萄糖和100μm本发明glut抑制剂化合物:(总体积200微升)。
上述空白对照组、阳性对照组、测试组各自分别设3孔,实验在37℃培养1小时。然后用冷却至零度的pbs(ph=7.2)洗涤细胞3次(1500r/min,4分钟),然后用5毫升含有10mm的tris-hcl缓冲液(ph8.0,购于solarbio)和0.5%的tritonx-100(购于sigma)的裂解缓冲液于80℃进行细胞裂解15分钟。裂解后细胞置于4℃经1500r/min离心20分钟。针对上清液进行2-脱氧葡萄糖吸收测试。被红细胞经glut1吸收的2-脱氧葡萄糖的细胞内浓度以2-脱氧葡萄糖-6-磷酸的形式采用2-脱氧葡萄糖细胞吸收试剂盒(2-deoxyglucose(2dg)uptakemeasurementkit,购于cosmobioco.ltd.,日本)进行测试。测试结果分别以空白对照组糖吸收为100%折算后如下表3:
表3:
阳性对照化合物细胞松弛素b是已知的glut1高活性抑制剂(procnatlacadsci.2016,113(17):4711-4716)。表3中结果显示,本发明化合物针对glut的2-脱氧葡萄糖吸收显示出了不同程度的抑制作用。
试验例3:化合物在抑制肿瘤生长中的作用
(1)细胞毒性测试:
细胞毒性实验采用mts测试方法。收集对数期肿瘤细胞,调整细胞悬液浓度,每孔加入100μl,铺板使待测细胞调密度至1000-10000个/孔,(边缘孔用无菌pbs填充)。在5%co2,37℃孵育,至细胞单层铺满孔底(96孔平底板),加入固定浓度的glut抑制剂化合物(50μm),每孔100μl,设5个复孔。在5%co2,37℃条件下孵育96小时,倒置显微镜下观察。向2mlmts(2mg/ml,dpbs配制)溶液中加入100μlpms(1mg/ml,dpbs配制),混匀,制成mts工作液。上述细胞培养板离心后弃去培养液,小心用pbs冲2-3遍后,在检测吸光度前,向96孔板中每孔加入100μl培养基,再加入20μlmts工作液,在37℃,5%co2条件下孵育2h后,在490nm处检测od值(光密度值)。
对照组:
在上述同样条件下不添加被测glut抑制剂化合物,最后取得肿瘤细胞在490nm处检测od值,作为细胞100%生存的对照。
上述每个样品的实验重复5组,取平均od值,与对照组比较计算细胞存活率。
(2)实验结果:
表4
如表4所示,本发明细胞糖转运通道抑制剂对乳腺癌以及肺癌细胞都具有不同强度的抑制作用。
试验例4:癌细胞高表达糖转运通道蛋白的测试实验
(1)westernblotting免疫印迹试验实验步骤:
1)、在100mm的培养细胞皿里培养细胞,控制每一盘的细胞总量及细胞密度相当。待细胞长满后,用600μl的ripa裂解液裂解(碧云天p0013b)。加6xloadingbuffer(莱宝科技d1010)在细胞裂解液中,于95℃水浴或金属浴孵育30min左右。
2)、sds-page凝胶电泳分析(分离胶浓度为10%或12%),每孔上样量约20μl。跑胶结束后,准备转膜。方法为半干转。转膜使用的buffer为transferbuffer(如下表1所示),使用的膜为pvdf膜(ge公司,使用前用甲醇浸泡),滤纸为bio-rad品牌的(转膜前用transferbuffer浸泡)。
3)、转膜实验:从下到上的顺序是:滤纸、pvdf膜、胶、滤纸。放置好后,用试管或者玻璃棒沿着一个方向将气泡赶出,一般剪裁的膜、滤纸都和整个分离胶的大小相当。之后,盖好仪器上面的盖子。转膜电压为20v,时间为35min-40min。
4)、转膜后,将膜取出,标记好正面(左上角剪掉一个小三角),然后用5%脱脂奶粉(tbstbuffer配置的)(奶粉品牌:索莱宝)室温孵育1h(20-50ml不等)。
5)、用tbstbuffer清洗pvdf膜,后按照marker的位置将膜剪开(目的蛋白和内参剪开),然后放在小盒子里分别孵育一抗。
6)、于4℃孵育一抗,过夜。配置一抗是用tbst+0.5%脱脂奶粉(5ml)。一抗浓度根据抗体说明书来定,一般用的浓度都是说明书上说的最低浓度,或比说明书的最低浓度偏高10倍。
7)、用tbst洗3次,时间分别是5min、10min、10min。
8)、于4℃孵育二抗,1h。配置一抗是用tbst+0.5%脱脂奶粉(5ml)。二抗浓度根据抗体说明书来定,一般用的浓度说明书的最低浓度。
9)、用tbst洗3次,时间分别是5min、10min、10min。
10)、用hrp酶标记的化学发光显色液显色(milliporewbkls0500),按照说明书操作,曝光拍照。
表5.10×transferbuffer
加ddh2o定容至800ml,充分搅拌。使用时,取80ml稀释至800ml,然后加20%甲醇(200ml)。
表6.10×tbstbuffer
加ddh2o溶解至约900ml后,调节ph为7.6,定容至1000ml。使用时稀释到1×,加0.05%tween20。
(2)实验结果:
实验结果如图4所示。实验结果显示,本发明glut抑制剂发挥肿瘤抑制作用中所使用的肺癌及乳腺癌与人体正常细胞相比均高表达糖转运通道蛋白。
实验证明,(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6,上述所列举本发明glut抑制剂化合物,能够针对细胞的糖转运通道glut,抑制其对糖分子的吸收。同时肿瘤细胞由于沃伯格效应高表达glut糖吸收通道蛋白,该类glut抑制剂对肿瘤细胞的生长产生抑制作用。
因此,本发明提供的所述细胞糖转运通道抑制剂可用于制备抗肿瘤药物。
作为抗肿瘤的药物组合物,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),可任选其一为唯一活性成分,其余是药学可接受的辅料,如赋形剂或稀释剂的混合。
作为抗肿瘤的药物组合物,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),的药学上可接受的盐为唯一活性成分。
作为抗肿瘤的药物组合物,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),的溶剂化物。
作为抗肿瘤的药物组合物,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),其余为药物载体。
作为糖转运通道蛋白glut抑制试剂,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),可任选其一为唯一活性成分,其余是药学可接受的辅料,如赋形剂或稀释剂的混合。
作为糖转运通道蛋白glut抑制试剂,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),的药学上可接受的盐为唯一活性成分。
作为糖转运通道蛋白glut抑制试剂,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),的溶剂化物。
作为糖转运通道蛋白glut抑制试剂,包括上述glut抑制剂化合物(分别为(i)-(a-1)-1、(i)-(a-2)-1、(i)-(a-3)-1、(i)-(a-1)-2、(i)-(a-2)-2、(i)-(a-3)-2、(i)-(a-1)-3、(i)-(a-2)-3、(i)-(a-3)-3、(i)-(a-1)-4、(i)-(a-2)-4、(i)-(a-3)-4、(i)-(a-1)-5、(i)-(a-2)-5、(i)-(a-3)-5、(i)-(a-1)-6、(i)-(a-2)-6、(i)-(a-3)-6),其余为药物载体。
上述药物组合物在制备抗肿瘤药物的用途。
上述化合物在制备细胞糖转运通道蛋白glut抑制试剂中的用途。
同时,作为glut抑制剂,通过阻断细胞的糖分子吸收,还能为研究包括肿瘤在内的细胞营养代谢相关的分子机制提供一种分子工具。
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明,对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!