一种具有1,3,4-恶二唑侧链的截短侧耳素衍生物及制备与应用
本发明属于药物化学领域,尤其涉及一种具有1,3,4-恶二唑侧链的截短侧耳素衍生物及制备与应用。
背景技术:
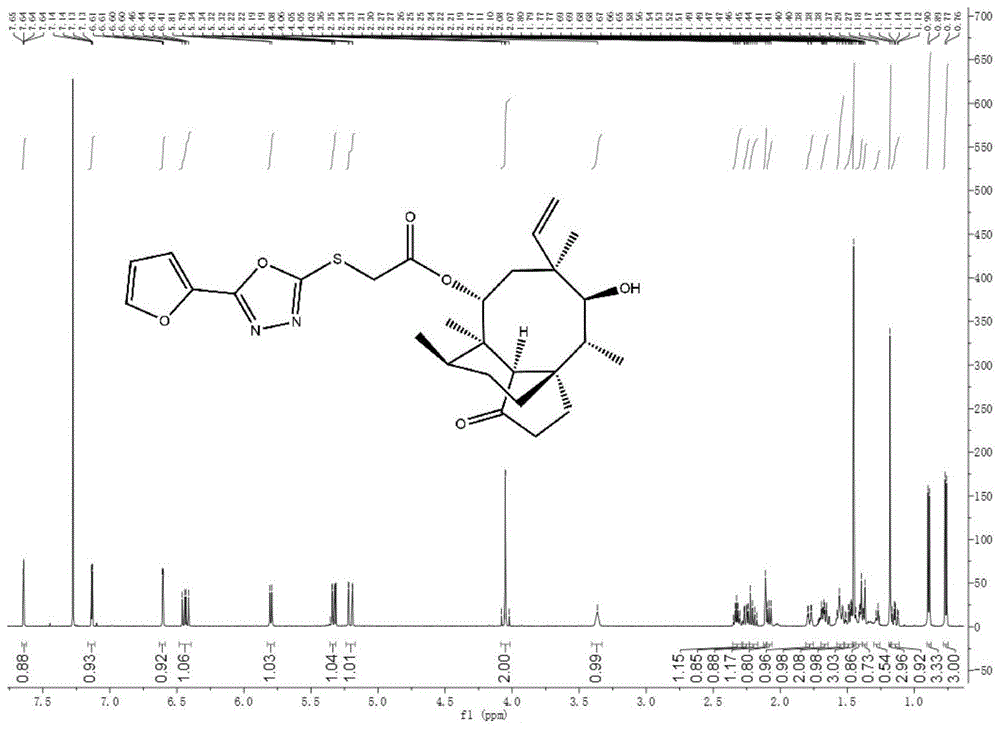
:作为高发病率和高死亡率的主要病原体之一,耐多药性金黄色葡萄球菌(mrsa)通常会引起与医疗保健相关的感染,对全世界的人类健康构成严重威胁。因此,迫切需要开发具有新作用方式的新抗生素,以用于治疗由多重耐药性金黄色葡萄球菌(mrsa)引起的感染。天然产物一直在应对人类疾病中发挥着重要作用,尤其是在传染病领域。尽管存在一些缺点,但已开发出多种天然产物或其衍生物,并被批准用作抗菌剂,例如氨曲南、万古霉素、替加环素和青霉素g。从天然产物或其半合成衍生物开发新的抗菌剂仍然是应对mrsa感染的最有效方法。截短侧耳素(pleuromutilin,式1)是一种天然的三环二萜类化合物,由侧耳菌pleurotusmutilus和pleurotuspasseckerianus培养产生,具有八个立体中心极具刚性的5-6-8三环碳骨架和c(14)的乙醇酸链等结构。研究表明,截短侧耳素类及其衍生物对革兰氏阳性菌显示出有效的抗菌活性,且有别与临床广泛应用的其他抗菌药物,截短侧耳素及其衍生物可通过与细菌50s核糖体亚基23srna的肽基转移酶中心(ptc)的v区结合而抑制细菌蛋白质的合成。由于独特的作用机理,截短侧耳素及其衍生物具有与其他抗菌剂之间交叉耐药性的低发生率以及细菌对细菌耐药性的低发展倾向,同时基本不与哺乳动物细胞核糖体发生相互作用,也不干扰真核细胞中的蛋白质合成。令研究人员注意的是,截短侧耳素结构分子中c14上的侧链,能够深入核糖体亚基的疏水基团内部,提高其抗菌活性。因此,通过化学手段来修饰c14侧链一直是提高截短侧耳素衍生物成药性的重要手段。到目前为止,通过改造其c14侧链,成功上市的抗菌药物已有兽用抗菌药泰妙菌素(tiamulin)、沃尼妙林(valnemulin),人皮肤外用药瑞它妙林(retapamulin)以及于2019年通过美国fda审批上市,用于治疗社区获得性细菌性肺炎(cabp)的人用药乐福妙林(lefamulin)共四款。与基于同一母核开发成功的药物,如青霉素、头孢菌素、沙星类抗菌药动辄数十种相比,截短侧耳素仅成功开发四种抗菌药,且针对截短侧耳素类抗菌药的耐药菌尚不多见。因此,开发更多截短侧耳素类抗菌药十分必要。技术实现要素:为了克服现有技术的不足和缺点,本发明的首要目的在于提供一种具有1,3,4-恶二唑侧链的截短侧耳素衍生物,此类截短侧耳素衍生物具有良好的抗菌活性,特别适合作为新型抗菌药物用于动物或人全身系统感染。本发明的另一目的在于提供上述具有1,3,4-恶二唑侧链的截短侧耳素衍生物的制备方法。本发明的再一目的在于提供上述具有1,3,4-恶二唑侧链的截短侧耳素衍生物的应用。本发明目的通过以下技术方案实现:一种具有1,3,4-恶二唑侧链的截短侧耳素衍生物,为式2化合物或其药学上可接受的盐,以及所述的式2化合物或其药学上可接受的盐的溶剂化合物、对映异构体、非对映异构体、互变异构体或其任意比例的混合物,包括外消旋混合物:其中,r为r1或r1为氢原子、呋喃基、噻吩基、苯基、2-吡啶基、3-吡啶基、4-吡啶基和吡嗪基中的一种;r2为甲基、三氟甲基、氨基、羟基、羟基、氢原子、氟原子、氯原子和溴原子中的一种;r3为甲基、硝基、氨基、氢原子、氟原子、氯原子和溴原子中的一种;r4为甲基、三氟甲基、氨基、二甲氨基、羟基、甲氧基、氢原子、氟原子、氯原子和溴原子中的一种;且r2、r3、r4不能同时为氢原子;优选的,所述r2为甲基,r3为氢原子,r4为氢原子;或者r2为氢原子,r3为甲基,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为甲基;或者r2为三氟甲基,r3为氢原子,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为三氟甲基;或者r2为氢原子,r3为氨基,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为氨基;或者r2为氢原子,r3为氢原子,r4为二甲氨基;或者r2为氢原子,r3为硝基,r4为氢原子;或者r2为羟基,r3为氢原子,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为羟基;或者r2为氢原子,r3为氢原子,r4为甲氧基;或者r2为氟原子,r3为氢原子,r4为氢原子;或者r2为氢原子,r3为氟原子,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为氟原子;或者r2为氯原子,r3为氢原子,r4为氢原子;或者r2为氢原子,r3为氯原子,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为氯原子;或者r2为氢原子,r3为溴原子,r4为氢原子;或者r2为氢原子,r3为氢原子,r4为溴原子;上述优选结构的化合物的具体基团归纳如表1所示:表1化合物编号及具体基团所述的药学上可接受的盐为式2化合物与盐酸、氢溴酸、硫酸、硝酸、磷酸、醋酸、富马酸、马来酸、草酸、丙二酸、琥珀酸、柠檬酸、苹果酸、甲磺酸、乙磺酸、苯磺酸、甲苯磺酸、谷氨酸或天冬氨酸形成的盐;所述的药学上可接受的盐优选具有如下结构式:所述的具有1,3,4-恶二唑侧链的截短侧耳素衍生物的制备方法,包含如下步骤:(1)将截短侧耳素与对甲基苯磺酰氯反应,得到如式3所示结构的中间体i;(2)将羧酸作为原料,与无水乙醇进行反应,得到如式4所示结构的中间体ii;(3)将步骤(2)制得的中间体ii与水合肼反应,得到如式5所示结构的中间体iii;(4)将步骤(3)制得的中间体iii与二硫化碳反应,得到如式所6示的中间iv(在本发明的实施例中对应化合物1c~28c);(5)将步骤(1)制得的中间体i为原料,通过碘化钠进一步活化,再与步(4)制得的中间体iv反应,得到如式2所示结构的具有1,3,4-恶二唑侧链的截短侧耳素衍生物;所述的中间体ⅰ、ⅱ、ⅲ和ⅳ分别具有式3~6结构式:步骤(1)中所述的对甲苯磺酰氯与截短侧耳素摩尔比优选为1.1:1;步骤(2)中所述的羧酸与无水乙醇的摩尔比优选为1:1.1;步骤(3)中所述的反应优选采用无水乙醇作为溶剂,中间体ii与水合肼的摩尔比优选为1:1,反应条件优选为25~40℃反应2~3h;步骤(4)中所述的反应的具体操作优选为:采用95%(v/v)乙醇为溶剂先溶解氢氧化钾,在冰浴下加入中间体ⅲ,再滴加二硫化碳,78~80℃加热回流3~4h;所述的95%(v/v)乙醇的用量优选为中间体ⅲ质量的5~20倍,氢氧化钾与中间体ⅲ的摩尔比优选为1.2:1,二硫化碳与中间体ⅲ摩尔比优选为1.1:1;步骤(5)中所述的活化的具体操作优选为:以乙腈作为溶剂先溶解中间体i,再加入碘化钠和碱,78℃加热回流1~3h;所述的乙腈的用量优选为中间体i质量的30~40倍,所述的碱与中间体i的摩尔比优选为2:1,所述的碘化钠的摩尔数优选为碱摩尔数的10%;所述的碱优选为氢氧化钠、氢氧化钾、氢氧化铯、碳酸钠、碳酸钾或碳酸铯;步骤(5)中所述的反应的条件优选为78~80℃加热回流1~3h;合成路线如下式所示:所述的具有1,3,4-恶二唑侧链的截短侧耳素衍生物在制备抗菌产品中的应用;所述的抗菌产品优选为治疗感染性疾病的药物;所述的抗菌产品进一步优选为用于治疗由革兰氏阳性菌引起的感染性疾病的抗菌药物;所述的感染性疾病为人或动物经耐药金黄色葡萄球菌或多药耐药菌感染引起的感染性疾病;所述的药物可含有一种或多种药学上可接受的载体、赋形剂或稀释剂;所述的药物的制剂包括多种临床药物剂型,如片剂、注射液、脂质体纳米粒、控释剂等;一种抗生素药物,含有有效量的具有1,3,4-恶二唑侧链的截短侧耳素衍生物,余量为药用辅料或其它可配伍的药物;所述药用辅料是指常规的药用赋形剂,如溶剂、崩解剂、矫味剂、防腐剂、着色剂和粘合剂等;所述其它可配伍的药物,指的是以有效剂量的具有1,3,4-恶二唑侧链的截短侧耳素衍生物为药物原料,再配伍其它天然药物或化学药品;与现有技术相比,本发明具有如下优点及有益效果:(1)本发明提供的截短侧耳素衍生物是未曾报道过的新类型化合物。(2)本发明经过广泛而深入的研究,合成了大量全新结构的具有1,3,4-恶二唑侧链的截短侧耳素衍生物,并进行了广泛的抗菌活性筛选,首次发现该类化合物不仅具有良好的体外抗菌活性,还具有较沃尼妙林(valnemulin)及瑞它莫林(retapamulin)制备成本低的优势,因而特别适合作为新型抗菌药物用于防治人或动物细菌感染性疾病,尤其是耐药金黄色葡萄球菌引起的感染性疾病。(3)本发明制得的具有1,3,4-恶二唑侧链的截短侧耳素衍生物水溶性好。附图说明图1是化合物2的核磁图谱图。图2是化合物3的核磁图谱图。图3是化合物15的核磁图谱图。图4是化合物19的核磁图谱图。具体实施方式下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。在实施例中,r-cooch2ch3中的r为r1或其中,具体基团r1、r2、r3、r4见表1,均为市购,其他试剂也均为市购。实施例1(1)中间体ⅰ制备:将10.0g(26.5mmol)截短侧耳素溶于20ml吡啶并置于冰浴;将5.6g(29.2mmol)对甲基苯磺酰氯溶于10ml吡啶,然后缓慢加入上述的截短侧耳素吡啶溶液,冰浴搅拌该混合液3h后,依次加入冰水与三氯甲烷各50ml,再转移至分液漏斗中振摇,静置待其分层;取有机相,依次用4mol/l的硫酸100ml、饱和碳酸氢钠溶液100ml、去离子水100ml洗涤;洗涤后有机相减压蒸发有机溶液,往剩余固体中加入异丙醇20ml,加热溶解后再放冷,析出大量白色粉末,抽滤,并用异丙醇洗涤滤渣,烘干,得到如式3所示结构的中间体ⅰ,产率81%;(2)中间体ii1a制备:取155.5mmol羧酸(r-cooch2ch3,其中,r为h)溶于10ml无水乙醇,冰浴搅拌该混合液3min后,缓慢加入0.15ml浓硫酸,115℃加热回流4h;将反应液倒入分液漏斗中,加30ml二氯甲烷萃取,并用去离子水洗涤两次,洗涤后用无水硫酸钠干燥,取有机相;所得有机相经过旋转蒸干,得到如式4所示结构的中间体ii1a,产率76.72%;(3)中间体iii1b制备:在机械搅拌下,向10ml无水乙醇中缓慢加入2.0g(40mmol)水合肼,再缓慢加入40mmol中间体ii,室温25℃反应2h后冰浴,析出大量固体,抽滤;将产物在真空干燥器中用cacl2干燥,最后用p2o5干燥,得到如式5所示结构的中间体iii1b,产率70.88%;(4)化合物1c(中间体iv-1)制备:取0.67g(12mmol)氢氧化钾溶于15ml的95%(v/v)乙醇,在冰浴条件下加入60.06mg(10mmol)中间体iii;将0.84g(11mmol)二硫化碳缓慢滴加上述反应体系中,滴加完毕后在80℃下条件下加热回流;加热回流3h后,在冰浴条件下,向反应体系中滴加浓盐酸,将反应体系ph值调为1,析出大量晶体,抽滤;将晶体在真空干燥器中用cacl2干燥,最后用p2o5干燥,得到如式6所示结构的化合物1c(中间体iv-1),产率81.17%。按照步骤(1)~(4)的方法(各反应物摩尔量、反应条件、纯化等均相同),分别得到中间体ii2a~中间体ii28a(产率75.55~88.64%)、中间体iii2b~中间体iii28b(产率62.84~74.25%)、化合物2c~化合物28c(产率75.35~86.54%),各中间体的产率见表2。实施例222-o-(1,3,4-恶二唑基)硫乙酰基妙林(化合物1)的制备取2.13g(4mmol)实施例1制得的中间体ⅰ溶于81ml乙腈,加入无水碘化钠0.12g(0.8mmol)和无水碳酸钾1.11g(8mmol),78℃加热回流反应2h,然后往上述体系加入0.45g(4.4mmol)实施例1制得的1,3,4-1,3,4-恶二唑-2-硫醇(中间体iv-1)78℃继续反应3h,将反应液倒入分液漏斗中,依次加入蒸馏水与三氯甲烷各50ml,振摇后静置待其分层,取其有机相再用氯化钠水溶液(15%w/v)洗涤两次并用无水硫酸钠干燥,取有机相;所得有机相旋转蒸干得混合物经二氯甲烷复溶,加入100~200目硅胶2g充分混合,待溶剂挥发完后,将上述粗产物-硅胶粉混合物用柱层析进行纯化(100~200目硅胶粉为固定相,二氯甲烷:甲醇=200:1(v:v)为流动相),得到22-o-(1,3,4-1,3,4-恶二唑基)硫乙酰基妙林的纯品,产率为73.74%。实施例3化合物2~28的制备按照实施例2相同的方法(各反应物摩尔量、反应条件、纯化等均与实施例2相同),仅将化合物1c(中间体iv-1)替换为化合物2c~28c(中间体iv-2~中间体iv-28),得到相应的式2所示产物,编号依次为2~28。其中,图1~4是化合物2、3、15和19的核磁图谱图。上述化合物制备产率归纳如表2所示。表2化合物编号及产率效果实施例(1)体外抑菌实验实验采用的是肉汤稀释法。实验对照药物选用泰妙菌素。泰妙菌素为截短侧耳素类抗生素,是世界十大兽用抗生素之一。实验中所用的菌株为耐甲氧西林金黄色葡萄球菌atcc43300和金黄色葡萄球菌atcc29213,临床金黄色葡萄球菌(staphylococcusaureus)ad3和临床金黄色葡萄球菌(s.aureus)144(临床菌ad3和144于华南农业大学兽医学院药理实验室分离鉴定,已在文献zheza,kangla,gyza,etal.design,synthesisandbiologicalactivitiesofnovelpleuromutilinderivativeswithasubstitutedtriazolemoietyaspotentantibacterialagents-sciencedirect[j].europeanjournalofmedicinalchemistry,2020.中公开)。目标化合物储备液配制:分别精密称取6.4mg目标化合物置于10ml容量瓶中,用0.25mldmso溶解,加入9.5ml蒸馏水,0.25ml吐温80定容,充分摇匀,得到储备液(6.4mg/ml),用0.22μm滤膜过除菌,小管分装,-20℃保存。对照药物泰妙菌素同样按照上述方法配制。菌液的配制:取出在-20℃下保存完好的菌株接种在新mh平板上,37℃培养24h后挑取单菌落接种在mh培养基中再次培养24h;选取单菌落转移到无菌的生理盐水中并调整其浊度为0.6mcf,此时菌液浓度为106cfu/ml。mic板制备:分别将目标化合物储备液(6.4mg/ml)稀释10倍,得到浓度为得到浓度为640μg/ml的目标化合物溶液;取无菌96孔板,第1孔加入180μlmh肉汤培养基,第2至10孔分别加入100μlmh肉汤培养基,往第1孔加入20μl浓度为浓度为640μg/ml的抗菌药物,混匀后取100μl加入第2孔,混匀,再吸取100μl至第3孔,依次类推,第12孔吸取100μl弃去。此时各孔药物浓度依次为:64、32、16、8、4、2、1、0.5、0.25、0.125、0.06、0.03μg/ml,每个浓度药物做三组平行。接种菌液:在1至12孔各加入100μl菌液,使每孔最终菌液浓度约为5×105cfu/ml,第1孔至第12孔药物浓度分别为32、16、8、4、2、1、0.5、0.25、0.125、0.06、0.03、0.015μg/ml。接种好的96孔板置于37℃培养箱进行培养,24h观察菌液生长情况。对照药物泰妙菌素同法测定以在小孔内完全抑制细菌生长的最低药物浓度为mic,阳性对照孔(即不含药物)内细菌需明显生长。当在微量肉汤稀释法出现单一跳孔时,记录抑制细菌的最高药物浓度,如出现多处跳孔则需重复试验。表3为mic结果,可知目标化合物对选用菌株具有良好的抑菌活性,具有良好的抑制耐药金黄色葡萄球菌活性,特别适合作为新型抗菌药物用于防治人或动物或耐药金葡或多药耐药菌引起的感染性疾病。表3体外抑菌数据(2)化合物溶解度的测定将化合物2、3、15和19形成硫酸盐,以瑞他妙林的硫酸盐为对照。采取高效液相色谱法测定其各自水中溶解度。试验结果见表4。表4化合物2、3、15、19以及瑞他妙林的硫酸盐溶解度化合物溶解度(mg/ml,ph=7.0水中)21.4231.57151.85192.32瑞他妙林(retapamulin)0.11由表4可知,测试的化合物都具有良好的水溶性,优于瑞他妙林盐的溶解性,改善了截短侧耳素类衍生物的溶解性,其中化合物19的硫酸酸盐水中溶解性达到2.32mg/ml。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1