减阻剂及其制备方法以及滑溜水压裂液和应用
本发明涉及压裂液
技术领域:
,具体涉及一种减阻剂及其制备方法以及滑溜水压裂液和应用。
背景技术:
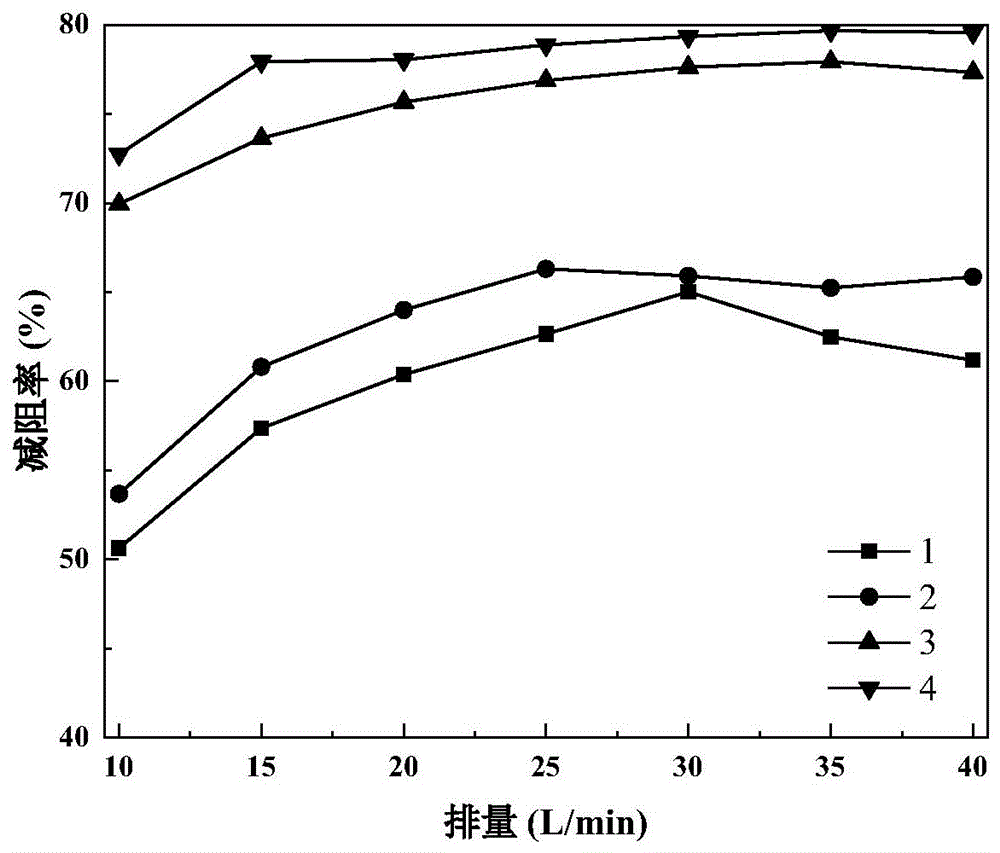
:石油作为一种高效能源在国民经济的发展中发挥着重大作用。超低渗、致密油藏孔隙结构复杂,储层物性较差,储层发育的多是微-纳米级孔喉,基质储量动用困难,常规水驱开发的采收率较低,多数油田采取压裂方式开发。压裂液的种类选择往往取决于油藏性质,油田较常用的水基压裂液体系主要有水基冻胶压裂液、胍胶压裂液、滑溜水压裂液三种。滑溜水压裂液以其低成本低伤害的特性广泛应用于非常规油气的开发。在大排量注入情况下,滑溜水中需要加入减阻剂,从而实现大排量泵注,弥补了水粘度低、携砂能力差等诸多问题。滑溜水压裂液技术是目前页岩气开发作业中应用最多的压裂液技术,其特征是以聚合物为主剂,配合杀菌剂、表面活性剂和阻垢剂等辅剂从而形成完整配方体系,具有较高的黏度和较好的减阻作用,与清水相比可减阻60%以上。目前有关滑溜水压裂液的研究主要在于减阻剂的优化,以配液简便的乳液型聚丙烯酰胺类减阻剂为例,虽然溶解速度快,但耐剪切效果差,在高排量下连续剪切20分钟减阻率就从75%下降到60%以下。常规粉末型纯聚丙烯酰胺类减阻剂可在高排量下达到70%甚至是75%以上的减阻率,但耐温效果较差,在超过90℃聚合物链剪切破坏严重,减阻率不足50%。近年来,随着非常规油气田资源的勘探和开发,对压裂增产技术要求越来越高,迫使国内外对滑溜水压裂液的研究也逐渐成熟,其种类不断增多。目前滑溜水压裂液受到减阻效果和耐温耐剪切效果等因素限制,现有技术中还没有比较成熟的能够兼顾减阻和耐温耐剪切的滑溜水压裂液。因此,研究和开发一种滑溜水压裂液具有重要意义。技术实现要素:本发明的目的是为了克服现有技术存在的滑溜水压裂液耐剪切性能和耐温性能的缺陷问题,提供一种减阻剂及其制备方法以及滑溜水压裂液和应用,该滑溜水压裂液兼具有良好的耐温和耐剪切性,并具有较好的减阻效果。为了实现上述目的,本发明第一方面提供了一种减阻剂,其中,所述减阻剂含有式(1)所示的结构单元和式(2)或式(3)所示的结构单元;其中,m为20000-28000,n1为90-130,n2为50-80;所述减阻剂的重均分子量为150万-200万。本发明第二方面提供了一种前述所述的减阻剂的制备方法,其中,所述的制备方法包括:(1)在溶剂存在下,将纳米二氧化硅和带双键的硅烷偶联剂接触进行第一反应,得到改性纳米二氧化硅;(2)在氮气和引发剂存在下,将所述改性纳米二氧化硅、助分散剂、式(4)所示的丙烯酰胺和水接触进行第二反应,以及将得到的反应产物进行洗涤、真空干燥和研磨处理,得到减阻剂;其中,所述带双键的硅烷偶联剂具有式(5)或式(6)所示的单体:其中,x1选自-cl和/或-ch3;其中,x2选自-cl、-ch3、-ch2ch3和-ch2ch2och3中的一种或多种。本发明第三方面提供了一种滑溜水压裂液,其中,所述滑溜水压裂液含有减阻剂、助排剂、黏土稳定剂和水,其中,所述减阻剂为前述所述的减阻剂。本发明第四方面提供了一种前述所述的滑溜水压裂液在油田压裂上的应用。通过上述技术方案,本发明具有以下优势:(1)本发明提供的减阻剂为纳米二氧化硅改性丙烯酰胺共聚物,与常规聚丙烯酰胺相比,在相同运动黏度下减阻率高20%以上,分散效果好,减阻性能优异,在浓度较低时就能够达到78%以上的减阻率,能够有效控制滑溜水压裂液的成本。(2)本发明提供的滑溜水压裂液能够有效地降低滑溜水体系表面张力(表面张力<28mn/m),提高润湿性,易于压裂液返排;同时能有效抑制黏土矿物的水化膨胀与脱落,毛细管吸收法测定cst<1.5,从而提高裂缝导流能力。(3)本发明提供的滑溜水压裂液耐温耐剪切性好,150℃下各组分的配伍性良好,无需絮凝、沉淀出现,减阻率依然在75%以上;40l/min的排量下剪切1h,依然保持75%以上的减阻率。附图说明图1是对比例1制备的减阻剂溶液和对比例6制备的滑溜水压裂液、实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的减阻实验的减阻率与排量的关系的示意图;图2是对比例1制备的减阻剂溶液和对比例6制备的滑溜水压裂液、实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的减阻实验的减阻率与剪切时间的关系的示意图;图3是对比例1制备的减阻剂溶液和对比例6制备的滑溜水压裂液、实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的减阻实验的减阻率与与温度的关系的示意图。附图标记说明1表示的是对比例1制备的减阻剂溶液;2表示的是对比例6制备的滑溜水压裂液;3表示的是实施例1制备的减阻剂溶液;4表示的是实施例7制备的滑溜水压裂液。具体实施方式在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。本发明第一方面提供了一种减阻剂,其中,所述减阻剂含有式(1)所示的结构单元和式(2)和/或式(3)所示的结构单元;其中,m为20000-28000,n1为90-130,n2为50-80;所述减阻剂的重均分子量为150万-200万。本发明的发明人发现:现有技术中纳米改性聚合物的合成主要通过三步发:(1)带氨基的硅烷偶联剂与二氧化硅反应;(2)改性的二氧化硅与顺丁烯二酸酐等带双键的酸酐类物质反应;(3)第二步的合成产物与am或其他的功能单体引发自由基聚合;但是,上述三步法合成的合成产物复杂且侧链较长,不利于减阻,所以没有用在滑溜水中。基于此,本发明的发明人意外发现,将带双键的硅烷偶联剂与纳米二氧化硅反应,得到改性二氧化硅;再将该改性二氧化硅与am引发自由基聚合,得到纳米二氧化硅改性聚丙烯酰胺共聚物,该共聚物的侧链更短,宜于体系减阻;另外,两步法能够进一步提供合成产率和效率。另外,在本发明中,选择带双键的硅烷偶联剂的优点是直接与丙烯酰胺单体在引发剂条件下引发自由基聚合,而如果选自带氨基的硅烷偶联剂,则会增加一步与带双键的酸酐类的中间产物,进而与丙烯酰胺单体在引发剂条件下引发自由基聚合,合成过程复杂,成本偏高。根据本发明,优选情况下,m为20900-27900,n1为100-120,n2为55-75。根据本发明,更优选情况下,所述减阻剂含有式(1)所示的结构单元和式(2)所示的结构单元时,m为21590-24955,n1为100-115。根据本发明,更优选情况下,所述减阻剂含有式(1)所示的结构单元和式(3)所示的结构单元时,m为21195-26920,n2为55-75。根据本发明,所述式(1)所示的结构单元和所述式(2)所示的结构单元的摩尔比为(185-250):1;所述式(1)所示的结构单元和所述式(3)所示的结构单元的摩尔比为(280-490):1。根据本发明,优选地,所述减阻剂的平均粒径为70-90nm,优选为75-85nm。根据本发明,所述纳米二氧化硅为粉末状,平均粒径为10-20nm。本发明第二方面提供了一种前述所述的减阻剂的制备方法,其中,所述的制备方法包括:(1)在溶剂存在下,将纳米二氧化硅和带双键的硅烷偶联剂接触进行第一反应,得到改性纳米二氧化硅;(2)在氮气和引发剂存在下,将所述改性纳米二氧化硅、助分散剂、式(4)所示的丙烯酰胺和水接触进行第二反应,以及将得到的反应产物进行洗涤、真空干燥和研磨处理,得到减阻剂;其中,所述带双键的硅烷偶联剂具有式(5)或式(6)所示的单体:其中,x1选自-cl和/或-ch3;其中,x2选自-cl、-ch3、-ch2ch3和-ch2ch2och3中的一种或多种。根据本发明,在步骤(1)中,所述溶剂为乙醇与水的混合体系,其中,以所述混合体系的总体积为基准,所述乙醇的体积为75-100%,所述水的体积为0-25%;优选地,乙醇:水=3:1。在本发明中,乙醇的浓度为99.99%,优选为无水乙醇。根据本发明,所述带双键的硅烷偶联剂选自甲基乙烯基二氯硅烷、乙烯基三乙氧基硅烷(ydh-151)、乙烯基三甲氧基硅烷(ydh-171)、乙烯基三(2-甲氧基乙氧基)硅烷(kh-172)和γ-甲基丙烯酰氧基丙基三甲氧基硅烷(kh-570)中的一种或多种;优选地,所述带双键的硅烷偶联剂选自乙烯基三乙氧基硅烷(ydh-151)、乙烯基三甲氧基硅烷(ydh-171)或γ-甲基丙烯酰氧基丙基三甲氧基硅烷(kh-570);更优选地,所述带双键的硅烷偶联剂为γ-甲基丙烯酰氧基丙基三甲氧基硅烷(kh-570)。根据本发明,所述溶剂、所述硅烷偶联剂和所述纳米二氧化硅的用量的重量比为(150-180):(0.05-1.5):(1.5-3),更优选为(150-165):(0.05-1):(2-3)。根据本发明,优选情况下,在步骤(1)中,将纳米二氧化硅先分散于一部分溶剂中,得到第一混合溶液;将带双键的硅烷偶联剂溶解于另一部分溶剂中,得到第二混合溶液;再将所述第一混合溶液和所述第二混合溶液混合。其中,所述混合的容器可以为三口烧瓶。另外,在本发明中,需要说明的是,所述溶剂为一部分溶剂和另一部分溶剂的总和。根据本发明,优选情况下,所述纳米二氧化硅与所述一部分溶剂的用量的重量比为(2-3):(80-90);所述带双键的硅烷偶联剂与所述另一部分溶剂的用量的重量比为(0.05-1):(70-75)。根据本发明,优选情况下,在步骤(1)中,所述的方法还包括:将所述纳米二氧化硅置于真空干燥箱中进行预处理,其中,所述预处理的条件包括:温度为120-130℃,时间为24-36h。根据本发明,优选情况下,在步骤(1)中,所述的方法还包括:将所述改性纳米二氧化硅进行旋蒸、洗涤和真空干燥处理;具体地,将所述改性纳米二氧化硅旋蒸后得到白色粉末状固体,用无水乙醇抽滤洗涤3-4次,在90-100℃下真空干燥24-36h,得到改性的纳米二氧化硅。根据本发明,在步骤(1)中,所述第一反应的条件包括:ph为3-4,搅拌速率为300-500r/min,温度为65-85℃,时间为3-4h;优选地,ph为3.5-4,搅拌速率为350-400r/min,温度为70-80℃,时间为3.5-4h;另外,在本发明中,可以选择1mol/l的草酸调节ph值。根据本发明,将纳米二氧化硅和带双键的硅烷偶联剂在三口烧瓶中进行接触,以及将该三口烧瓶置于油浴条件下进行所述第一反应。根据本发明,在步骤(2)中,所述引发剂选自过硫酸铵、亚硫酸氢钠和过硫酸钾中的一种或多种,优选地,所述引发剂选自过硫酸铵和亚硫酸氢钠,或者,所述引发剂选自过硫酸钾和亚硫酸氢钠;并且,更优选地,所述过硫酸铵和所述亚硫酸氢钠的用量的质量比为1:1;所述过硫酸钾和所述亚硫酸氢钠的用量的质量比为1:1。根据本发明,所述引发剂与所述改性纳米二氧化硅的用量的重量比为(0.03-0.05):(0.24-0.3),优选为(0.0375-0.04):(0.248-0.28)。根据本发明,所述助分散剂为乙醇。根据本发明,以丙烯酰胺、所述改性的纳米二氧化硅、所述助分散剂和水的总重量为基准,所述丙烯酰胺的用量为24-30重量%,所述改性纳米二氧化硅的用量为0.24-0.3重量%、所述助分散剂的用量为2-5重量%,水的用量为64.7-73.76重量%;优选情况下,以丙烯酰胺、所述改性的纳米二氧化硅、所述助分散剂和水的总重量为基准,所述丙烯酰胺的用量为24-26重量%,所述改性纳米二氧化硅的用量为0.25-0.28重量%、所述助分散剂的用量为2-3重量%,水的用量为70.72-75.75重量%。根据本发明,优选情况下,先将所述改性纳米二氧化硅溶于所述助分散剂和一部分水中,得到第一混合溶液;再将所述丙烯酰胺溶解于另一部分水中,得到第二混合溶液;然后将第一混合溶液和第二混合溶液混合,得到混合液,在该混合液中通入氮气1-2h后再加入引发剂。根据本发明,在步骤(2)中,所述第二反应的条件包括:ph为7-7.5,搅拌速率为150-250r/min,温度为30-40℃,时间为3-5h;优选地,搅拌速率为150-200r/min,温度为35-40℃,时间为3-4h。根据本发明,在步骤(2)中,将得到的反应产物进行洗涤、真空干燥和研磨处理,具体地,待产物固化后取出,切成小块,用无水乙醇洗涤3-4次,60-70℃下真空干燥48-60h,搅拌机粉碎即得到纳米二氧化硅改性的聚合物,即,减阻剂。本发明第三方面提供了一种滑溜水压裂液,其中,所述滑溜水压裂液含有减阻剂、助排剂、黏土稳定剂和水,其中,所述减阻剂为前述所述的减阻剂。根据本发明,以所述滑溜水压裂液的总重量为基准,所述减阻剂的含量为0.05-0.15重量%,所述助排剂的含量为0.05-0.5重量%,所述黏土稳定剂的含量为0.05-1重量%,水的含量为98.35-99.85重量%;优选地,以所述滑溜水压裂液的总重量为基准,所述减阻剂的含量为0.08-0.12重量%,所述助排剂的含量为0.1-0.3重量%,所述黏土稳定剂的含量为0.1-0.5重量%,水的含量为99.08-99.72重量%。在本发明中,将所述减阻剂、所述助排剂、所述黏土稳定剂和水的含量限定为前述范围之内,优点是在保持较高减阻率的同时能降低滑溜水压裂液的界面张力,使压裂液易于返排,也能有效抑制黏土矿物的水化膨胀。根据本发明,所述助排剂选自壬基酚聚氧乙烯醚np-10、十二醇聚氧乙烯醚和异构十醇聚氧乙烯醚中的一种或多种;优选地,所述助排剂为十二醇聚氧乙烯醚。根据本发明,所述黏土稳定剂为环氧氯丙烷-二乙醇胺共聚物和/或氯化铵;优选地,所述黏土稳定剂为环氧氯丙烷-二乙醇胺共聚物。本发明第四方面提供了一种前述所述的滑溜水压裂液在油田压裂上的应用。根据本发明,在10mm管径、40l/min的排量下0.08%的减阻剂溶液减阻率可达78%以上。根据本发明一种特别优选的实施方式,本发明提供的减阻剂的制备方法包括:(1)将粉末状的纳米二氧化硅在置于120-130℃真空干燥箱内干燥24-36h;取2-3g干燥后的纳米二氧化硅于烧杯中,加入80-90g溶剂无水乙醇,在室温下搅拌分散12h。另取0.05-1g硅烷偶联剂烷于烧杯,加入70-75g溶剂无水乙醇,在室温下搅拌至澄清。将两种溶液混合后置于三口烧瓶,边搅拌边用1mol/l的草酸调节ph为3.5-4,将混合液置于70-80℃油浴下搅拌反应3.5-4h。将反应产物旋蒸后得到白色粉末状固体,用无水乙醇抽滤洗涤3-4次,90-100℃下真空干燥24-36h,得到改性的纳米二氧化硅;2)以丙烯酰胺、改性的纳米二氧化硅、助分散剂无水乙醇和水的总重量为基准,称取24-26重量%的丙烯酰胺、0.25-0.28重量%的改性的纳米二氧化硅,2-3重量%无水乙醇,余量为水。先将改性的纳米二氧化硅溶于无水乙醇中,加入一定量的水,室温下搅拌分散至均匀,然后将丙烯酰胺、剩余的水,在搅拌下,ph为7-7.5,加热到35-40℃,然后通入氮气0.5-1h后加入0.0375-0.04%的引发剂,在氮气氛围下反应3-4h,至产物固化后取出,切成小块,用无水乙醇洗涤3-4次,60-70℃下真空干燥48-60h,粉碎即得到纳米二氧化硅改性的聚合物,即,减阻剂j1。以下将通过实施例对本发明进行详细描述。以下实施例和对比例中:减阻率参数通过jhif-2减阻仪的测量的差压计算得到;实施例1本实施例在于说明采用本发明的方法制备的减阻剂。(1)将2.5g干燥后的粒径为10-20nm的纳米二氧化硅溶于85g无水乙醇,在室温下搅拌分散12h,得到第一混合溶液。另取0.525g硅烷偶联剂γ-甲基丙烯酰氧基三甲氧基硅烷kh-570溶于72.5g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为375r/min条件搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,用1mol/l的草酸调节混合液的ph=3.75,将混合液置于75℃油浴下搅拌反应3.75h;并将反应产物在65℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为95℃条件真空干燥30h,即得到kh-570改性的纳米二氧化硅;(2)将2.5g助分散剂乙醇加入0.25g改性纳米二氧化硅,搅拌均匀后加入35g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取25g丙烯酰胺加入到39.75g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下,ph为7,加热到40℃,然后在该混合液中通入氮气1h后加入0.039g的引发剂(过硫酸铵和亚硫酸氢钠各0.0195g),在氮气氛围下反应3-4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为65℃条件真空干燥54h,冷却至室温后研磨,即得到纳米二氧化硅改性的聚合物,即,减阻剂j1;其中,该减阻剂j1含有式(1)所示的结构单元和式(3)所示的结构单元;并且,m为26900-26920,n2为72-75;所述减阻剂j1的重均分子量为193万,平均粒径为78.56nm。实施例2本实施例在于说明采用本发明的方法制备的减阻剂。(1)将2g干燥后的粒径为10-20nm的纳米二氧化硅溶于80g无水乙醇中,在室温下搅拌分散12h,得到第一混合溶液。另取0.05g硅烷偶联剂乙烯基三乙氧基硅烷溶于70g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为350r/min条件下搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,用1mol/l的草酸调节混合液的ph=4,将混合液置于80℃油浴下搅拌反应4h;并将反应产物在60℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为90℃条件真空干燥24h,即得到乙烯基三乙氧基硅烷改性的纳米二氧化硅;(2)将2g助分散剂乙醇加入0.25g改性纳米二氧化硅,搅拌均匀后加入35g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取24g丙烯酰胺加入到38.75g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下,ph为7.5,加热到30℃,然后在该混合液中通入氮气1h后加入0.0375g的引发剂(过硫酸铵和亚硫酸氢钠各0.01875g),在氮气氛围下反应3-4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为60℃条件真空干燥48h,冷却至室温后研磨,即得到纳米二氧化硅改性的聚合物,即,减阻剂j2;其中,该减阻剂j2含有式(1)所示的结构单元和式(2)所示的结构单元;并且,m为24945-24955,n1为112-115;所述减阻剂j2的重均分子量为179万,平均粒径为76.32nm。实施例3本实施例在于说明采用本发明的方法制备的减阻剂。(1)将3g干燥后的粒径为10-20nm的纳米二氧化硅溶于90g无水乙醇中,在室温下搅拌分散12h,得到第一混合溶液。另取1g乙烯基三甲氧基硅烷溶于75g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为400r/min条件搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,用1mol/l的草酸调节混合液的ph=4,将混合液置于80℃油浴下搅拌反应4h;并将反应产物在70℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为100℃条件真空干燥36h,即得到乙烯基三甲氧基硅烷改性的纳米二氧化硅;(2)将3g助分散剂乙醇加入0.28g改性纳米二氧化硅,搅拌均匀后加入35g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取26g丙烯酰胺加入到35.72g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下,ph为7.2,加热到40℃,然后在该混合液中通入氮气1h后加入0.04g的引发剂(过硫酸铵和亚硫酸氢钠各0.02g),在氮气氛围下反应3-4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为70℃条件真空干燥60h,冷却至室温后研磨,即得到纳米二氧化硅改性的聚合物,即,减阻剂j3;其中,该减阻剂j3含有式(1)所示的结构单元和式(2)所示的结构单元;并且,m为21590-21600,n1为100-105;所述减阻剂j3的重均分子量为155万,平均粒径为75.76nm。实施例4本实施例在于说明采用本发明的方法制备的减阻剂。(1)将2.5g干燥后的粒径为10-20nm的纳米二氧化硅溶于90g无水乙醇,在室温下搅拌分散12h,得到第一混合溶液。另取0.775g硅烷偶联剂γ-甲基丙烯酰氧基三甲氧基硅烷kh-570溶于75g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为350r/min条件搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,用1mol/l的草酸调节混合液的ph=3.75,将混合液置于75℃油浴下搅拌反应4h;并将反应产物在65℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为95℃条件真空干燥30h,即得到kh-570改性的纳米二氧化硅;(2)将3.5g助分散剂乙醇加入0.27g改性纳米二氧化硅,搅拌均匀后加入35g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取27g丙烯酰胺加入到34.23g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下,ph为7.3,加热到35℃,然后在该混合液中通入氮气1h后加入0.04g的引发剂(过硫酸铵和亚硫酸氢钠各0.02g),在氮气氛围下反应3-4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为65℃条件真空干燥54h,冷却至室温后研磨,即得到纳米二氧化硅改性的聚合物,即,减阻剂j4;其中,该减阻剂j4含有式(1)所示的结构单元和式(3)所示的结构单元;并且,m为22725-22735,n2为60-63;所述减阻剂j4的重均分子量为163万,平均粒径为79.76nm。实施例5本实施例在于说明采用本发明的方法制备的减阻剂。(1)将1.5g干燥后的粒径为10-20nm的纳米二氧化硅溶于80g无水乙醇中,在室温下搅拌分散12h,得到第一混合溶液。另取0.05g硅烷偶联剂γ-甲基丙烯酰氧基三甲氧基硅烷kh-570溶于70g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为350r/min条件搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,用1mol/l的草酸调节混合液的ph=4,将混合液置于65℃油浴下搅拌反应4h;并将反应产物在60℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为90℃条件真空干燥24h,即得到kh-570改性的纳米二氧化硅;(2)将2g助分散剂乙醇加入0.24g改性纳米二氧化硅,搅拌均匀后加入35g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取24g丙烯酰胺加入到38.76g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下,ph为7.5,加热到30℃,然后在该混合液中通入氮气1h后加入0.03g的引发剂(过硫酸铵和亚硫酸氢钠各0.015g),在氮气氛围下反应3-4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为60℃条件真空干燥48h,冷却至室温后研磨,即得到纳米二氧化硅改性的聚合物,即,减阻剂j5;其中,该减阻剂j5含有式(1)所示的结构单元和式(3)所示的结构单元;并且,m为23140-23150,n2为62-65;所述减阻剂j5的重均分子量为166万,平均粒径为82.12nm。实施例6本实施例在于说明采用本发明的方法制备的减阻剂。(1)将3g干燥后的粒径为10-20nm的纳米二氧化硅溶于95g无水乙醇,在室温下搅拌分散12h,得到第一混合溶液。另取1g硅烷偶联剂γ-甲基丙烯酰氧基三甲氧基硅烷kh-570溶于85g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为350r/min条件搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,用1mol/l的草酸调节混合液的ph=4,将混合液置于85℃油浴下搅拌反应4h;并将反应产物在60℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为100℃条件真空干燥24h,即得到kh-570改性的纳米二氧化硅;(2)将5g助分散剂乙醇加入0.3g改性纳米二氧化硅,搅拌均匀后加入30g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取30g丙烯酰胺加入到34.7g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下,ph为7.1,加热到40℃,然后在该混合液中通入氮气1h后加入0.05g的引发剂(过硫酸铵和亚硫酸氢钠各0.025g),在氮气氛围下反应3-4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为70℃条件真空干燥60h,冷却至室温后研磨,即得到纳米二氧化硅改性的聚合物,即,减阻剂j6;其中,该减阻剂j6含有式(1)所示的结构单元和式(3)所示的结构单元;并且,m为21195-21205,n2为55-57;所述减阻剂j6的重均分子量为152万,平均粒径为87.54nm。对比例1将2g干燥后的粒径为10-20nm的纳米二氧化硅溶于80g无水乙醇,在室温下搅拌分散12h,得到第一混合溶液。另取1.2g硅烷偶联剂3-氨基丙基三乙氧基硅烷kh-550溶于72g溶剂无水乙醇中,在温度为25℃条件下,在搅拌速率为350rpm条件搅拌至澄清,得到第二混合溶液。将第一混合溶液和第二混合溶液混合得到混合液,再加入1.825g三乙胺,将混合液置于80℃油浴下搅拌反应4h;并将反应产物在60℃条件下旋蒸、用无水乙醇洗涤3-4次、在温度为90℃条件真空干燥24h,即得到kh-550改性的纳米二氧化硅。称取1gkh-550改性的纳米二氧化硅,加入dmf做溶剂,在25℃条件下将上述溶液滴加至含有0.4g顺丁烯二酸酐以及20gdmf的溶液中,滴加时间为3h,滴加完后在70℃下反应12h,产物用去离子水洗涤,然后在70℃下真空干燥24,纳米二氧化硅表面已成功引入双键。将0.20g助分散剂乙醇加入0.248g改性纳米二氧化硅,搅拌均匀后加入35g水,室温下机械搅拌分散至澄清,得到第一混合溶液。另取24.752g丙烯酰胺加入到40g水中,得到第二混合溶液。将第一混合溶液和第二混合溶液混合,得到混合液,在搅拌下加热到40℃,然后在该混合液中通入氮气1h后加入0.05g的引发剂(过硫酸铵和亚硫酸氢钠各0.025g),在氮气氛围下反应3~4h,至产物固化后取出、切块、用无水乙醇洗涤3-4次、在温度为60℃条件真空干燥48h,在常温常压条件下研磨,即得到纳米二氧化硅改性的聚合物,结果制备得到减阻剂dj1。对比例2按照与实施例1相同的方法制备减阻剂,所不同之处在于:第二步反应温度为50℃。结果制备得到减阻剂dj2。对比例3按照与实施例1相同的方法制备减阻剂,所不同之处在于:第二步反应引发剂加量为0.08g的引发剂(过硫酸铵和亚硫酸氢钠各0.04g)。结果制备得到减阻剂dj3。对比例4按照与实施例1相同的方法制备减阻剂,所不同之处在于:第一步反应ph为6。结果制备得到减阻剂dj4。对比例5按照与实施例1相同的方法制备减阻剂,所不同之处在于:第二步反应丙烯酰胺的用量为25重量%,改性纳米二氧化硅的用量为0.49重量%,所述助分散剂的用量为3.5重量%,水的用量为74.51重量%。结果制备得到减阻剂dj5。实施例7本实施例在于说明采用本发明的方法制备的滑溜水压裂液。将0.09g实施例1制得的减阻剂加入99.56g水,在温度为25℃条件下,在搅拌速率为300r/min下机械搅拌24h后加入0.25g十二醇聚氧乙烯醚和0.1g环氧氯丙烷-二乙醇胺共聚物,在搅拌速率为250r/min手动搅拌后至分散均匀,即可得到滑溜水压裂液s7。实施例8-12本实施例在于说明采用本发明的方法制备的滑溜水压裂液。按照与实施例7相同的方法制备滑溜水压裂液,所不同之处在于:将“实施例1制备的减阻剂”分别替换为“实施例2-6制备的减阻剂”。结果分别得到滑溜水压裂液s8-s12。对比例6-10按照与实施例7的方法制备滑溜水压裂液,所不同之处在于:将“实施例1制备的减阻剂”分别替换为“对比例1-5制备的减阻剂”。结构制备得到滑溜水压裂液ds6-ds10。测试例1利用型jhif-2减阻仪评价实施例1-6制备的减阻剂溶液j1-j6和对比例1-5制备的减阻剂溶液d1-d6、实施例7-12制备的滑溜水压裂液s7-s12和对比例6-10制备的溜水压裂液ds6-ds10减阻效果。方法包括:管径为10mm,温度为25℃,记录清水和以上溶液的随排量变化差压。计算随排量变化的最高减阻率,结果如表1(减阻效果)所示。图1是对比例1制备的减阻剂溶液和对比例6制备的滑溜水压裂液、实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的减阻实验的减阻率与排量的关系的示意图;从图1能够看出:置于d1和sd1的最高减阻率为65.01%和66.32%,而j1和s7的最高减阻率分别为77.93%和79.57%,这表明实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液都有较好的减阻效果,其中s7的减阻效果最好。测试例2利用jhif-2型减阻仪评价实施例1-6制备的减阻剂溶液j1-j6和对比例1-5制备的减阻剂溶液d1-d6、实施例7-12制备的滑溜水压裂液s7-s12和对比例6-10制备的溜水压裂液ds6-ds10耐剪切性。方法包括:管径为10mm,温度为25℃,排量40l/min,记录清水和以上溶液的随时间变化差压。计算随剪切1h后的减阻率的下降值,结果如表1(耐剪切性)所示。图2是对比例1制备的减阻剂溶液和对比例6制备的滑溜水压裂液、实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的减阻实验的减阻率与剪切时间的关系的示意图;从图2能够看出:置于d1和sd1的减阻率从65%以上降低到55%以下,而j1的减阻率仅从78.88%降低到75.01%,s1制得的滑溜水压裂液的减阻率几乎没有变化,这表明实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液都较好的耐剪切性,其中滑溜水压裂液的耐剪切性效果最好。测试例3利用jhif-2型减阻仪评价实施例1-6制备的减阻剂溶液j1-j6和对比例1-5制备的减阻剂溶液d1-d6、实施例7-12制备的滑溜水压裂液s7-s12和对比例6-10制备的溜水压裂液ds6-ds10耐温性。方法包括:管径为10mm,温度25-150℃,排量40l/min,记录清水和以上三种溶液的随温度变化差压。计算随温度从25℃升高到150℃的减阻率下降值,结果如表1(耐温性)所示。图3是对比例1制备的减阻剂溶液和对比例6制备的滑溜水压裂液、实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的减阻实验的减阻率与温度的关系的示意图。从图3能够看出:置于d1和sd1的减阻率降低到50%以下,而j1和s1的减阻率几乎没有变化,这表明实施例1制备的减阻剂溶液和实施例7制备的滑溜水压裂液的耐温性明显优于d1和sd1,其中滑溜水压裂液的耐温性效果最好。表1实施例编号减阻效果耐剪切性耐温性实施例177.9%3.8%5.2%实施例276.8%4.2%5.5%实施例377.1%4.4%6.2%实施例476.5%6.7%8.9%实施例576.2%7.5%10.2%实施例675.4%6.9%9.3%实施例779.6%2.4%6.1%实施例878.2%2.7%7.1%实施例978.7%3.1%6.3%实施例1077.2%4.5%9.7%实施例1176.9%8.6%11.1%实施例1275.6%8.0%10.2%对比例171.0%11.2%19.5%对比例268.3%9.3%10.8%对比例357.3%15.7%23.9%对比例466.6%16.6%17.7%对比例552.6%2.8%2.3%对比例666.3%10.4%21.2%对比例769.2%14.9%12.1%对比例856.1%15.1%26.7%对比例967.5%3.7%5.3%对比例1052.2%3.6%4.2%备注:减阻效果以减阻率为评价指标,耐剪切性和耐温性以减阻率的减少量(增加为负,减小为正)为评价指标。通过表1的结果可以看出,采用本发明的实施例具有耐温耐剪切和减阻明显更好的效果。对比例1是采用硅烷偶联剂3-氨基丙基三乙氧基硅烷kh-550制备的减阻剂,由于纳米二氧化硅接枝率低,因此,耐温耐剪切效果不好。对比例2是在制备过程中,改变了聚合反应的温度,由于反应中后期以后,引发剂残留量很少,聚合度较低,减阻剂的分子量较小,因此,耐温耐剪切和减阻效果不好。对比例3是在制备过程中,改变了引发剂加量,由于初级自由基数量过少,单体转化率较低,减阻剂的分子量较小,因此,耐温耐剪切和减阻效果不好。对比例4是在制备过程中,改变了反应ph,由于体系呈明显的酸性,单体最终转化率不高,减阻剂的分子量较小,因此,耐温耐剪切和减阻效果不好。对比例5是在制备过程中,改变了单体配比,由于二氧化硅浓度过高,影响了聚合产物链长增长,减阻剂的分子量较小,因此,减阻效果不好。对比例6-10由于没有采用本发明制备的减阻剂,合成的聚合物分子量普遍较小,分子链长较短,因此,减阻效果不好。以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1