一种抗病毒化合物及其制备方法与流程
1.本发明属于医药领域,具体涉及一种抗病毒化合物及其制备方法。
背景技术:
2.流行性感冒病毒简称流感病毒。它分为甲(a)、乙(b)、丙(c)三型,近年来才发现的流感病毒将归为丁(d)型。流感病毒可引起人、禽、猪、马、蝙蝠等多种动物感染和发病,是人流感、禽流感、猪流感、马流感等人与动物疫病的病原。
3.这些疫病典型的临床症状是急性高热、全身疼痛、显著乏力和呼吸道症状。流感病毒主要通过空气中的飞沫、易感者与感染者之间的接触或与被污染物品的接触而传播。一般秋冬季节是其高发期。人流感主要是甲型流感病毒和乙型流感病毒引起的。甲型流感病毒经常发生抗原变异,可以进一步分为h1n1、h3n2、h5n1、h7n9等亚型(其中的h和n分别代表流感病毒两种表面糖蛋白)。流感病毒对外界抵抗力不强。动物流感病毒通常不感染人,人流感病毒通常不感染动物,但是猪比较例外。猪既可以感染人流感病毒,也可以感染禽流感病毒,但它们主要感染的还是猪流感病毒。少数动物流感病毒适应人后,可以引起人流感大流行。
4.人冠状病毒可对人造成普通感冒,严重急性呼吸综合征(sars)和中东呼吸综合征(mers),在流行病学特征上存在一定差异。
5.在全球,10%~30%的上呼吸道感染由hcov
‑
229e、hcov
‑
oc43、hcov
‑
nl63和hcov
‑
hku1四类冠状病毒引起,在造成普通感冒的病因中占第二位,仅次于鼻病毒。感染呈现季节性流行,每年春季和冬季为疾病高发期。潜伏期2
‑
5天,人群普遍易感。主要通过人与人接触传播。sars由人感染sars
‑
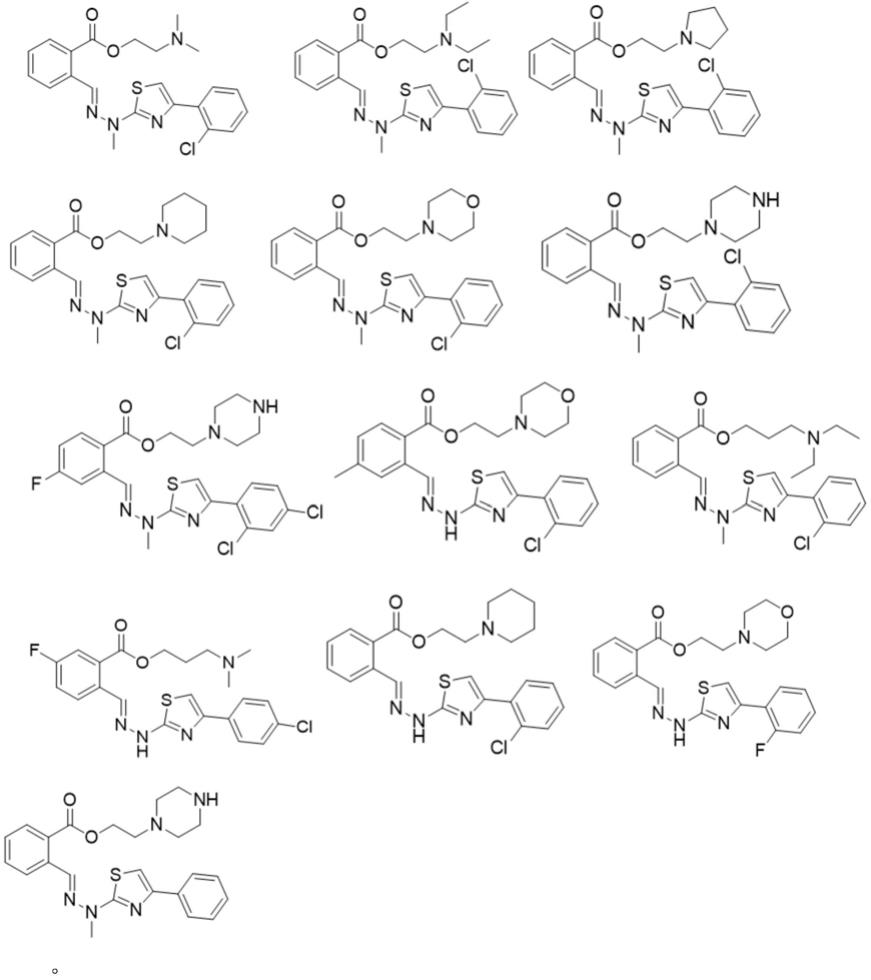
cov引起,首先出现在我国广东省部分地区,之后波及我国24个省、自治区、直辖市和全球其他28个国家和地区。2002年11月至2003年7月全球首次sars流行中,全球共报告临床诊断病例8096例,死亡774例,病死率9.6%。sars的潜伏期通常限于2周之内,一般约2~10天。人群普遍易感。sars病人为最主要的传染源,症状明显的病人传染性较强,潜伏期或治愈的病人不具备传染性。mers是一种由mers
‑
cov引起的病毒性呼吸道疾病,于2012年在沙特阿拉伯首次得到确认。自2012年起,mers在全球共波及中东、亚洲、欧洲等27个国家和地区,80%的病例来自沙特阿拉伯,病死率约35%。潜伏期最长为14天,人群普遍易感。单峰骆驼是mers
‑
cov的一大宿主,且为人间病例的主要传染来源,人与人之间传播能力有限。
6.甲型h1n1流感是猪的一种高度传染性急性呼吸道疾病,由一种或多种猪流感a型病毒引起。发病率往往较高,但死亡率较低(1~4%)。通过浮质、直接和间接接触以及携带病毒但无症状的猪,病毒在猪群中传播。全年都可发生猪群疫情。在温带的秋季和冬季,发病率上升。人患甲型h1n1流感通常来自被感染的猪,但有些人患病例没有与猪或猪所在环境接触的历史。在有些情况中发生了人际传播,但仅限于密切接触者和封闭环境中的人群。
7.冠状病毒(hcov
‑
229e)是冠状病毒的一种。冠状病毒属于套式病毒目、冠状病毒科、冠状病毒属,是一个大型病毒家族,在自然界广泛存在。冠状病毒仅感染脊椎动物,与人
和动物的多种疾病有关,可引起人和动物呼吸道、消化道和神经系统疾病。
8.因此,研究一种有效的抗病毒药物具有重要的现实意义。
技术实现要素:
9.本发明的目的是提供一种式i所示的化合物及其药学上可接受的盐、溶剂合物。
[0010][0011]
所述式(ⅰ)中,n=0
‑
5的整数,如1、2、3;
[0012]
r1选自:单取代或多取代的h、f、甲基、三氟甲基,优选h;
[0013]
r2选自:h、直链或取代烷烃(c1
‑
c6),优选甲基,异丙基;
[0014]
r3选自:单取代或多取代的h、cl、br、f,优选cl;
[0015]
r4、r5独立地选自下述基团中的任意一种:(c1‑
c6)烷基、(c3‑
c8)碳环基烷基、含有取代基的(c1‑
c
18
)的烷烃、(c2‑
c8)烯基、含有取代基的(c2‑
c8)烯基、(c2‑
c8)炔基、含有取代基的(c2‑
c8)炔基、(c6‑
c
20
)芳基、含有取代基的(c6‑
c
20
)芳基、(c2‑
c
20
)杂环基、含有取代基的(c2‑
c
20
)杂环基;或r4、r5相互成环为(c3‑
c8)杂环烷基或取代(c3‑
c8)杂环烷基、(c6‑
c
20
)杂芳基或取代(c6‑
c
20
)杂芳基。
[0016]
所述r4、r5中所述的取代基选自下述至少一种:甲基,乙基,异丙基,吡咯基,哌啶基,吗啉基和哌嗪基。
[0017]
所述(c3‑
c8)杂环烷基具体可为吡咯基,哌啶基,吗啉基或哌嗪基。
[0018]
在本发明的一些实施方案中,本发明所述的式i所示化合物可以列举为如下所示结构,但不局限于以下结构:
[0019][0020]
药效学试验表明,通式(ⅰ)中化合物当r4和r5均不为h时,能够获得较高的代谢物zonk2003
‑
0的血药浓度,具有优良的生物利用度,auc远高于灌胃等摩尔的zonk2003
‑
0时的血药浓度,而当r4取代基为h时如zonk2003
‑
22,则灌胃后获得非常低的zonk2003
‑
0的血药浓度,提示具有非常低的生物利用度或者转变成了其它物质。
[0021]
本发明还提供了上述式i所示的化合物的制备方法。
[0022]
根据文献med.chem.commun.,2016,7,1441
–
1448,化合物1(式a)烷基肼与硫氰酸钠反应得到硫脲化合物2(式b),然后与r1取代的邻甲酸苯甲醛缩合得到化合物3(式c),最后与r3取代溴苯乙酮关环,很容易得到噻唑类化合物4(式d);在缩合剂作用下,得到式i所示的苯甲酰胺衍生物。具体步骤如下:
[0023]
1)将式a所示化合物与硫氰酸钠进行反应,得到式b所示化合物;
[0024][0025]
其中,式a中r2的定义同式i;式b中r2的定义同式a;
[0026]
2)将式b所示化合物与式e所示的r1取代的邻甲酸苯甲醛进行缩合反应,得到式c所示化合物;
[0027][0028]
其中,式e中r1的定义同式i;式c中r1的定义同式e、r2的定义同式b;
[0029]
3)将式c所示化合物与式f所示的r3取代溴苯乙酮进行关环反应,得到式d所示化合物;
[0030][0031]
其中,式f中r3的定义同式i,式d中r1、r2的定义同式c,r3的定义同式f;
[0032]
4)在缩合剂作用下,使式d所示化合物与式g所示化合物进行缩合反应,得到式i所示化合物;
[0033][0034]
其中,式g中n、r4、r5的定义同式i。
[0035]
上述步骤1)中,所述反应的反应条件为:反应温度50
‑
100℃,反应时间为24
‑
72小时;反应在溶剂中进行,所述溶剂可为甲醇、乙醇、四氢呋喃、乙腈等,优选乙醇。
[0036]
上述步骤2)中,所述缩合反应的反应条件为:反应温度50
‑
100℃,反应时间为1
‑
3小时;反应在溶剂中进行,所述溶剂可为甲醇、乙醇、四氢呋喃、乙腈等,优选乙醇。
[0037]
上述步骤3)中,所述关环反应的反应条件为:反应温度50
‑
100℃,反应时间为3
‑
6小时;反应在溶剂中进行,所述溶剂可为甲醇、乙醇、四氢呋喃、乙腈等,优选乙醇。
[0038]
上述步骤4)中,所述缩合反应的反应条件为:反应温度0
‑
25℃,反应时间为2
‑
8小时;反应在溶剂中进行,所述溶剂可为二氯甲烷、四氢呋喃、乙腈等,优选二氯甲烷;。
[0039]
参照本发明实施例的制备方法可以获得本发明权利要求中所保护的其它化合物。
[0040]
本发明另一个目的是提供上述式i所示化合物的应用。
[0041]
本发明所提供的应用是式i所示化合物或其药学上可接受的盐、酯、溶剂合物的应用为下述(a)和/或(b)和/或(c):
[0042]
(a)式i所示化合物或其药学上可接受的盐、酯、溶剂合物在制备治疗病毒所致疾病或病毒感染的产品中的应用;
[0043]
(b)式i所示化合物或其药学上可接受的盐、酯、溶剂合物在制备预防病毒所致疾病或病毒感染的产品中的应用;
[0044]
(c)式i所示化合物或其药学上可接受的盐、酯、溶剂合物在制备病毒抑制剂中的应用。
[0045]
所述产品可为药物或药物制剂。
[0046]
所述病毒抑制剂能够抑制病毒的复制。
[0047]
所述病毒包括流感病毒、冠状病毒。
[0048]
所述流感病毒具体可为甲型流感病毒(h1n1);
[0049]
所述冠状病毒可为α属冠状病毒和/或β属冠状病毒,具体选自hcov
‑
229e。
[0050]
本发明中,所述病毒所致疾病可为呼吸系统感染性疾病。
[0051]
所述呼吸系统感染为呼吸道感染和/或肺部感染;所述呼吸道感染可为鼻咽炎、鼻炎、咽喉炎、气管炎和/或支气管炎;所述肺部感染可为肺炎。
[0052]
本发明中,所述流感病毒所致疾病通常包括流感病毒引起的急性呼吸道传染疾病等。
[0053]
本发明中,所述冠状病毒所致疾病通常包括病毒性肺炎、严重急性呼吸综合征等。
[0054]
本发明中,所述冠状病毒感染通常引起病毒性肺炎、严重急性呼吸综合征等疾病。
[0055]
本发明化合物同时具有对冠状病毒和h1n1甲型流感病毒的抑制作用,并且对人正常细胞没有毒性,能够抑制炎症反应发生的程度,减小肺炎对机体的伤害,促进机体恢复。
[0056]
以式i所示的化合物为活性成分制备的抗病毒药物也属于本发明的保护范围。
[0057]
所述抗病毒药物可通过注射、喷射、滴鼻、滴眼、渗透、吸收、物理或化学介导的方法导入机体如肌肉、皮内、皮下、静脉、粘膜组织;或是被其他物质混合或包裹后导入机体。
[0058]
需要的时候,在上述药物中还可以加入一种或多种药学上可接受的载体。所述载体包括药学领域常规的稀释剂、赋形剂、填充剂、粘合剂、湿润剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂等。
[0059]
上述药物可以制成片剂、粉剂、颗粒剂、胶囊、口服液、膏剂、霜剂、注射液等多种形式;上述各种剂型的药物均可以按照药学领域的常规方法制备。
[0060]
本发明还提供了一种药物或药物组合物,其活性成分为式i所示化合物或其药学上可接受的盐、酯、溶剂合物。
[0061]
所述药物或药物组合物具有下述至少一种功效:
[0062]
1)治疗病毒所致疾病或病毒感染;
[0063]
2)预防病毒所致疾病或病毒感染;
[0064]
3)抑制病毒。
[0065]
上述药物或药物组合物可以按照本领域技术人员已知的常规方法制成溶液剂、片剂、胶囊或注射剂等剂型。
[0066]
利用本发明提供的式i所示化合物或其药学上可接受的盐预防和/或治疗病毒引
起的感染时,给予受试者生物体有效量的式i化合物或其药学上可接受的盐。
[0067]
本发明中所述化合物经过实验证实,不仅对于h1n1甲型流感病毒具有较好的抑制作用,并且对于冠状病毒也具有较好的抑制作用,没有观察到对于人正常细胞的毒性,且能够在抗病毒的同时抑制炎症反应的程度。
附图说明
[0068]
图1为本发明式i所示化合物的合成路线图。
具体实施方式
[0069]
下面结合具体实施例对本发明作进一步阐述,但本发明并不限于以下实施例。所述方法如无特别说明均为常规方法。所述原材料如无特别说明均能从公开商业途径获得。
[0070]
实施例1
‑
10
[0071]
1、(e)
‑2‑
(甲氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
22)
[0072]
1)2
‑
甲基氨基硫脲(1
‑
2)
[0073][0074]
将甲基肼(23.0g,0.5mol)、硫氰酸铵(38.0g,0.5mol)、乙醇(200ml)分别加入单口圆底烧瓶中,加热回流反应72h。反应液冷却浓缩,柱层析纯化得到灰白色固体2
‑
甲基氨基硫脲(44.1g,84.0%)。1h nmr(dmso
‑
d
6 400mhz)δ7.24(s,2h),6.85(s,2h),3.14(s,3h).esi
‑
ms m/z:106.1[m+h]
+
.
[0075]
2)(e)
‑2‑
((2
‑
氨基甲硫杂酰
‑2‑
甲基亚肼基)甲基)苯甲酸(1
‑
3)
[0076][0077]
将2
‑
甲基氨基硫脲(40.0g,0.38mol)、2
‑
羰基苯甲酸(57.0g,0.38mol)、乙醇(300ml)分别加入单口圆底烧瓶中,加热回流反应2h。反应液冷却浓缩,柱层析纯化得到浅黄色固体(e)
‑2‑
((2
‑
氨基甲硫杂酰
‑2‑
甲基亚肼基)甲基)苯甲酸(85.6g,95.0%)。1h nmr(dmso
‑
d
6 400mhz)δ13.0(s,1h),8.12
‑
7.23(m,7h),2.47(s,3h).esi
‑
ms m/z:238.1[m+h]
+
.
[0078]
3)(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(1
‑
4)(zonk2003
‑
0)
[0079]
[0080]
将(e)
‑2‑
((2
‑
氨基甲硫杂酰
‑2‑
甲基亚肼基)甲基)苯甲酸(80.0g,0.34mol)、2
‑
溴
‑1‑
(2
‑
氯苯基)乙酮(79.2g,0.34mol)、乙醇(400ml)分别加入单口圆底烧瓶中,加热回流反应3h。反应液冷却浓缩,柱层析纯化得到浅黄色固体(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(124g,98.5%)。1h nmr(dmso
‑
d
6 400mhz)δ13.23(s,1h),8.60(s,1h),8.01
‑
7.34(m,9h),3.66(s,3h).esi
‑
ms m/z:372.1[m+h]
+
.
[0081]
4)(e)
‑2‑
((叔丁氧羰基)(甲基)氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(1
‑
5)
[0082][0083]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、二环己基碳二亚胺(2.27g,11mmol),室温搅拌3h。tlc检测原料消失后,加入2
‑
(n
‑
boc
‑
n
‑
甲基氨基)乙醇(1.75g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
((叔丁氧羰基)(甲基)氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(4.52g,85.5%)。1h nmr(dmso
‑
d
6 400mhz)δ8.24(s,1h),8.00
‑
7.34(m,9h),4.68(m,2h),3.64(s,3h),3.42(m,2h),3.26(s,3h),1.36(s,9h).esi
‑
ms m/z:529.5[m+h]
+
.
[0084]
5)(e)
‑2‑
(甲氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成
[0085][0086]
将(e)
‑2‑
((叔丁氧羰基)(甲基)氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(4.52g,8.54mmol)溶解于二氯甲烷(20ml)中,加入浓盐酸(10ml),回流反应3h。tlc检测原料消失后,反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
(甲氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(3.22g,88.1%)。1h nmr(dmso
‑
d
6 400mhz)δ8.24(s,1h),8.00
‑
7.34(m,9h),4.68(m,2h),3.64(s,3h),3.42(m,2h),3.26(s,3h).esi
‑
ms m/z:429.5[m+h]
+
.
[0087]
2、(e)
‑2‑
(二甲基氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
23)
[0088][0089]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳酰二亚胺盐酸盐(2.11g,11mmol),室温搅拌3h。tlc检测原料消失后,加入2
‑
(二甲基氨基)乙醇(0.89g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
(二甲基氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(3.68g,83.3%)。1h nmr(dmso
‑
d6400mhz)δ8.20(s,1h),8.01
‑
7.34(m,9h),4.55(m,2h),4.07(m,2h),3.52(s,3h),2.68(s,6h).esi
‑
ms m/z:443.5[m+h]
+
.
[0090]
3、(e)
‑2‑
(二乙氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
9)
[0091][0092]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、二环己基碳二亚胺(2.27g,11mmol),室温搅拌3h。tlc检测原料消失后,加入2
‑
(二乙基氨基)乙醇(1.17g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
(二乙氨基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(3.85g,81.8%)。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,9h),4.55(m,2h),3.52(s,3h),3.24
‑
3.01(m,6h),1.12(m,6h).esi
‑
ms m/z:471.1[m+h]
+
.
[0093]
实施例4、(e)
‑2‑
(吡咯烷
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
10)
[0094][0095]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳酰二亚胺盐酸盐(2.11g,11mmol),室温搅拌3h。tlc检测原料消失后,加入2
‑
(吡咯烷
‑1‑
基)乙醇盐酸盐(1.52g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
(吡咯烷
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(3.64g,77.6%)。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,9h),4.55(m,2h),3.52(s,3h),3.24
‑
3.01(m,6h),1.68(m,4h).esi
‑
ms m/z:469.5[m+h]
+
.
[0096]
实施例5、(e)
‑2‑
(哌啶
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
24)
[0097][0098]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、二环己基碳二亚胺(2.27g,11mmol),室温搅拌3h。tlc检测原料消失后,加入n
‑
羟乙基哌啶(1.29g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
(哌啶
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)甲基亚肼基)甲基)苯甲酸酯(3.86g,80.1%)。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,9h),4.55
‑
3.54(m,11h),1.86
‑
1.55(m,6h).esi
‑
ms m/z:483.5[m+h]+.
[0099]
6、(e)
‑2‑
吗啉乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
25)
[0100][0101]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、1
‑
乙基
‑
(3
‑
二甲基氨基丙基)碳酰二亚胺盐酸盐(2.11g,11mmol),室温搅拌3h。tlc检测原料消失后,加入2
‑
吗啉乙醇(1.31g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
吗啉乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)甲基亚肼基)甲基)苯甲酸酯(3.70g,76.5%)。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,9h),4.55
‑
3.54(m,15h).esi
‑
ms m/z:485.5[m+h]
+
.
[0102]
实施例7、(e)
‑2‑
(哌嗪
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
26)
[0103][0104]
将(e)
‑2‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸(3.71g,10mmol)溶解于二氯甲烷(30ml)中,分别加入n
‑
羟基丁二酰亚胺(1.15g,10mmol)、二环己基碳二亚胺(2.27g,11mmol),室温搅拌3h。tlc检测原料消失后,加入1
‑
哌嗪乙醇(1.30g,10mmol),继续室温反应5h。反应液浓缩,柱层析纯化得到类白色固体(e)
‑2‑
(哌嗪
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯(3.58g,74.1%)。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.11(m,9h),4.55
‑
3.54(m,12h),3.31(s,3h).esi
‑
ms m/z:484.5[m+h]
+
.
[0105]
实施例8、(e)
‑2‑
(哌嗪
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2,4
‑
二氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)
‑4‑
氟苯甲酸酯的合成(zonk2003
‑
27)
[0106][0107]
综合实施例7的制备方法,合成得到(e)
‑2‑
(哌嗪
‑1‑
基)乙基2
‑
((2
‑
(4
‑
(2,4
‑
二氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)
‑4‑
氟苯甲酸酯。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.11(m,7h),4.55
‑
3.54(m,12h),3.31(s,3h).esi
‑
ms m/z:536.5[m+h]
+
.
[0108]
实施例9、(e)
‑2‑
吗啉乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)亚肼基)甲基)
‑4‑
甲基苯甲酸酯的合成(zonk2003
‑
28)
[0109][0110]
综合实施例6的制备方法,合成得到(e)
‑2‑
吗啉乙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)亚肼基)甲基)
‑4‑
甲基苯甲酸酯。1h nmr(dmso
‑
d
6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,8h),4.55
‑
3.54(m,12h),2.68(s,3h).esi
‑
ms m/z:485.5[m+h]+.
[0111]
实施例10、(e)
‑3‑
(二乙基氨基)丙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
29)
[0112][0113]
综合实施例3的制备方法,合成得到(e)
‑3‑
(二乙基氨基)丙基2
‑
((2
‑
(4
‑
(2
‑
氯苯基)噻唑
‑2‑
基)
‑2‑
甲基亚肼基)甲基)苯甲酸酯1h nmr(dmso
‑
d6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,9h),4.55(m,2h),3.52(s,3h),3.24
‑
3.01(m,6h),1.68(m,2h),1.12(m,6h).esi
‑
ms m/z:485.5[m+h]
+
.
[0114]
实施例11、(e)
‑3‑
(二甲氨基)丙基2
‑
((2
‑
(4
‑
(4
‑
氯苯基)噻唑
‑2‑
基)亚肼基)甲基)
‑5‑
氟苯甲酸酯的合成(zonk2003
‑
8)
[0115][0116]
综合实施例3的制备方法,合成得到(e)
‑3‑
(二甲氨基)丙基2
‑
((2
‑
(4
‑
(4
‑
氯苯基)噻唑
‑2‑
基)亚肼基)甲基)
‑5‑
氟苯甲酸酯。1h nmr(dmso
‑
d6 400mhz)δ8.20(s,1h),8.01
‑
7.34(m,8h),4.55(m,2h),3.52(m,2h),3.24(s,6h),1.86(m,2h).esi
‑
ms m/z:461.1[m+h]+.
[0117]
实施例12、(e)
‑2‑
(哌嗪
‑1‑
基)乙基2
‑
((2
‑
甲基
‑2‑
(4
‑
苯基噻唑
‑2‑
基)亚肼基)甲基)苯甲酸酯的合成(zonk2003
‑
13)
[0118][0119]
综合实施例3的制备方法,合成得到(e)
‑2‑
(哌嗪
‑1‑
基)乙基2
‑
((2
‑
甲基
‑2‑
(4
‑
苯基噻唑
‑2‑
基)亚肼基)甲基)苯甲酸酯。1h nmr(dmso
‑
d6 400mhz)δ8.20(s,1h),8.01
‑
7.11(m,10h),4.55
‑
3.54(m,12h),3.31(s,3h).esi
‑
ms m/z:450.1[m+h]+.
[0120]
实施例13、系列化合物对甲型流感病毒a/fm/1/47(h1n1)感染小鼠的保护作用
[0121]
受试物:
[0122]
zonk2003
‑
0;zonk2003
‑
22;zonk2003
‑
9;zonk2003
‑
25;及zonk2003
‑
23、zonk2003
‑
10、zonk2003
‑
24、zonk2003
‑
26、zonk2003
‑
27、zonk2003
‑
28、zonk2003
‑
8,由广东中科药物研究有限公司提供。
[0123]
磷酸奥司他韦颗粒,规格15mg
×
10袋装,宜昌东阳光长江药业股份有限公司产品,批号:0371912115,有效期至2021.12.11,用于抗甲型流感病毒的阳性对照药;
[0124]
实验材料:甲型流感病毒鼠肺适应株a/fm/1/47(h1n1),接种鸡胚,收集尿囊液保
存。icr小鼠,体重18~22g。给药期间自由进食、饮水,每天12小时光照,12小时黑暗,温度22
±
2℃,湿度55~70%。实验方法:适应性饲养3天后,开始进行实验。除未感染对照组以外,其它各组小鼠用乙醚轻度麻醉,鼻腔内接种用生理盐水稀释的相当于8
×
ld
50
的流感病毒a/fm/1/47(h1n1)的鸡胚尿囊液50μl/只,阳性对照奥司他韦组和实施例化合物组小鼠于感染后2h首次灌胃给药,每种化合物以10μmol/kg、20μmol/kg、30μmol/kg的剂量灌胃给药,每天给药两次,连续用药5天。观察14天内小鼠的存活情况,
[0125]
并计算药物对于小鼠的死亡保护率(死亡保护率=模型组死亡率一实验组死亡率)。
[0126]
表1、化合物对a型流感病毒(h1n1甲型流感病毒)感染小鼠的保护作用
[0127][0128][0129]
实施例14:药物对流感病毒h1n1感染导致的小鼠肺部炎症的缓解作用
[0130]
实验方法:适应性饲养3天后,开始进行实验。除未感染对照组以外,其它各组小鼠用乙醚轻度麻醉,鼻腔内接种用生理盐水稀释的相当于8
×
ld
50
的流感病毒a/fm/1/47(h1n1)的鸡胚尿囊液50μl/只,阳性对照奥司他韦组小鼠和供试给药组于病毒感染24h后以80mg/kg首次灌胃给药,以后每日1次,病毒对照组及未感染对照组同法口服生理盐水,每日1次,给药体积为0.1ml/10g体重。共5天。第6天每组取3只小鼠称重,摘除眼球放血致死,取出全肺,称重,计算肺指数及肺指数抑制率。
[0131]
失重百分比%=给药前体重
‑
给药后体重/给药前体重
×
100%
[0132]
肺指数=小鼠肺重/小鼠体重
×
100
[0133]
肺指数抑制率(%)=病毒对照组肺指数均数
‑
给药组肺指数均数/病毒对照组肺指数均数
×
100%
[0134]
组别失重百分比%肺指数肺指数抑制率%正常对照组 0.57 模型对照组 2.09 奥司他韦组15.121.6620.57zonk2003
‑
89.781.2540.19zonk2003
‑
912.451.1943.06zonk2003
‑
1011.021.0848.32
[0135]
实验结果:因此实施例化合物对流感病毒引起的肺部炎症有明显的保护和抑制作用,且效果优于奥司他韦对照组。
[0136]
实施例15:zonk2003药物灌胃大鼠药代动力学实验
[0137]
雄性大鼠每组含3只,分别为zonk2003
‑
0组、zonk2003
‑
22组、zonk2003
‑
9组、zonk2003
‑
25组,灌胃,每只大鼠采集10个不连续的时间点。
[0138]
建立测定icr小鼠全血中zonk2003
‑
0、zonk2003
‑
22、zonk2003
‑
9、zonk2003
‑
25浓度的lc
‑
ms/ms分析方法。所得血药浓度数据同时采用药动学处理软件pharsight phoenix winnonlin 8.0非房室模型计算相关药代动力学参数。
[0139]
大鼠灌胃zonk2003
‑
0,剂量为2.51mg/kg;
[0140]
大鼠灌胃等摩尔的zonk2003
‑
22,剂量为3.14mg/kg;
[0141]
大鼠灌胃等摩尔的zonk2003
‑
9,剂量为3.27mg/kg;
[0142]
大鼠灌胃等摩尔的zonk2003
‑
25,剂量为3.42mg/kg;
[0143]
溶剂:20%solutol hs
‑
15/生理盐水;
[0144]
检测zonk2003
‑
0的血药浓度,计算药代动力学参数。
[0145]
详细结果见下表。
[0146]
表2大鼠灌胃给药2.51mg/kg的zonk2003
‑
0后,大鼠体内原型药物血药浓度检测结果(ng/ml)
[0147][0148]
表3大鼠灌胃给药2.51mg/kg的zonk2003
‑
0后,原型药物主要药代动力学参数
[0149][0150]
表4大鼠灌胃给药3.14mg/kg的zonk2003
‑
22后,代谢物zonk2003
‑
0血药浓度检测结果(ng/ml)
[0151][0152]
nd:未检测到,即血药浓度达峰后测定值低于定量下限点,—:该值无法计算
[0153]
表5大鼠灌胃给药3.14mg/kg的zonk2003
‑
22后,代谢物zonk2003
‑
0主要药代动力学参数
[0154]
[0155]
表6大鼠灌胃给药3.27mg/kg的zonk2003
‑
9后,代谢物zonk2003
‑
0血药浓度检测结果(ng/ml)
[0156][0157][0158]
表7大鼠灌胃给药3.27mg/kg的zonk2003
‑
9后,代谢物zonk2003
‑
0主要药代动力学参数
[0159][0160]
表8大鼠灌胃给药3.42mg/kg的zonk2003
‑
25后,代谢物zonk2003
‑
0血药浓度检测结果(ng/ml)
[0161][0162]
表9大鼠灌胃给药3.42mg/kg的zonk2003
‑
25后,代谢物zonk2003
‑
0主要药代动力学参数
[0163][0164][0165]
灌胃等摩尔的zonk2003
‑
9和zonk2003
‑
25后,检测到代谢物zonk2003
‑
0的血药浓度,并且auc为灌胃等摩尔zonk2003
‑
0的2
‑
3倍,提示zonk2003
‑
9和zonk2003
‑
25的生物利用度是zonk2003
‑
0灌胃的2
‑
3倍,zonk2003
‑
9和zonk2003
‑
25是zonk2003
‑
0的优良的前体药物,而灌胃等摩尔的zonk2003
‑
22后检测代谢物zonk2003
‑
0的血药浓度,auc远低于zonk2003
‑
9和zonk2003
‑
5组,也低于zonk2003
‑
0灌胃组,提示zonk2003
‑
22灌胃后生物利用度低或者是转变成了其它物质,未能获得较高的代谢物zonk2003
‑
0的血药浓度。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1