一种从草果中提取分离草果素的方法与流程
1.本发明是一种从草果中提取分离草果素的方法,具体涉及一种从草果中提取分离抗氧化活性物质草果素的方法,属于中草药提取技术领域。
背景技术:
2.草果(学名:amomum tsaoko crevost et lemarie)是姜科,豆蔻属多年生草本植物,主要分布于中国云南、广西、贵州等省区。草果是药食两用中的药材大宗品种之一,主要作为香料应用于食品加工领域,也应用于中药和其他领域。目前,随着天然产物开发的不断兴起,草果作为重要的药食两用植物,由于具备广泛的生物学活性和药理作用,因此,对草果天然产物的开发利用将会进一步加大。
3.现有研究表明,草果的提取物具有一定的抗氧化活性,例如:黄比翼等在“不同极性溶剂的草果提取物抗氧化活性研究”(《右江名族医学院学报》,2021年2月,第43卷 第1期,37
‑
40)中用乙醇提取草果得到的浸膏,稀释后依次用石油醚、乙酸乙酯、正丁醇萃取,得到石油醚部位、乙酸乙酯部位、正丁醇部位以及水部位,并采用紫外分光光度法来测定草果提取物对abts
+
自由基、羟自由基和fe
2+
螯合的清除能力,结果表明,不同极性溶剂的草果提取物都具有良好的抗氧化活性。但现有研究并未阐明其抗氧化活性的物质基础,因此,为进一步的提高草果在抗氧化活性方面的综合开发及利用,如何实现草果提取物中抗氧化活性单体化合物的提取、分离和纯化是关键。
技术实现要素:
4.本发明的目的在于提供一种从草果中提取分离草果素的方法,可实现草果药材中抗氧化活性单体化合物的提取,并经ms、nmr等现代波谱技术和光谱技术对该活性单体化合物进行结构鉴定,确认为草果素,由此为研究草果水提取物抗氧化活性提供物质基础,并有利于草果的进一步综合开发利用。
5.本发明通过下述技术方案实现:一种从草果中提取分离草果素的方法,包括以下步骤:a.以草果药材为原料,粉碎后用乙醇回流提取,再经减压浓缩后得到浓缩液;b.将浓缩液稀释后用乙酸乙酯进行萃取,得到乙酸乙酯提取物,浓缩制得乙酸乙酯提取物浸膏;c.取乙酸乙酯提取物浸膏,经硅胶柱层析分离和c18柱反向分离后,得到草果素。
6.所述步骤a中,用质量浓度为95%的乙醇回流提取2~3次,每次回流提取时间控制在1.5~2h。
7.所述步骤a中,将回流提取得到的提取液在50~55℃下减压浓缩至无醇后,得到所述浓缩液。
8.所述步骤b中,将浓缩液加水稀释2~3倍后,取等体积的乙酸乙酯萃取2次,得到乙酸乙酯提取物。
9.所述步骤b中,将乙酸乙酯提取物在45℃下减压浓缩制得乙酸乙酯提取物浸膏。
10.所述步骤c中,取乙酸乙酯提取物浸膏用其质量1~2倍的硅胶拌样,拌样物在40℃下鼓风干燥恒重,得到干燥的硅胶拌样物,再进行硅胶柱层析分离。
11.所述步骤c中,硅胶拌样物进行正相硅胶柱层析,以10~20倍的硅胶为填料,湿法装柱,干法上样,用体积比为100︰0~30︰1的二氯甲烷
‑
甲醇为洗脱剂进行常压洗脱,分管收集,每管约100ml,再进行薄层色谱分析,以体积比为20︰1的二氯甲烷
‑
甲醇为展开剂,在254nm的紫外光下检视暗斑结果,将含有目标化合物的收集液合并后,在45℃下减压浓缩至浸膏。
12.所述步骤c中,c18柱反向分离时,采用体积比为50︰50的甲醇
‑
水为流动相,紫外光为254nm。
13.本发明与现有技术相比,具有以下优点及有益效果:(1)本发明提供了一种从草果中提取抗氧化活性单体化合物的方法,通过乙醇回流提取浓缩液经乙酸乙酯萃取得到草果抗氧化活性物质的乙酸乙酯提取物,再经分离、纯化后得即可得到活性单体化合物。
14.(2)本发明为研究草果提取物的抗氧化活性提供了物质基础,经ms、nmr等现代波谱技术和光谱技术对本发明提取得到的活性单体化合物进行结构鉴定,确定该活性单体化合物为草果素。
15.(3)本发明为进一步实现草果的综合开发和利用奠定了基础,在对本发明提取得到的活性单体化合物草果素进行抗氧化活性实验时,依次通过dpph自由基清除实验、abts自由基清除实验、超氧阴离子自由基清除实验、羟基自由基清除实验、亚铁离子的螯合实验、三价铁离子的还原实验发现,草果素对这些自由基及铁离子均具有一定的清除能力,说明草果素具有一定的抗氧化能力,其抗氧化活性与单体化合物结构有关,抗氧化作用机理主要是通过清除自由基来避免氧化损伤。
16.综上所述,本发明在现有草果抗氧化活性研究的基础上,首次提出了从草果中提取活性单体化合物的方法,并对其提取得到的活性单体化合物进行鉴定和实验验证,充分证明了草果中抗氧化活性物质草果素的抗氧化性能,为草果的进一步综合开发利用提供研究基础。
附图说明
17.图1为单体化合物ⅰ的紫外光谱图。
18.图2为单体化合物ⅰ的质谱图。
19.图3为单体化合物ⅰ对dpph自由基的清除能力图。
20.图4为单体化合物ⅰ对abts自由基的清除能力图。
21.图5为单体化合物ⅰ对超氧阴离子自由基的清除能力图。
22.图6为单体化合物ⅰ对羟基自由基的清除能力图。
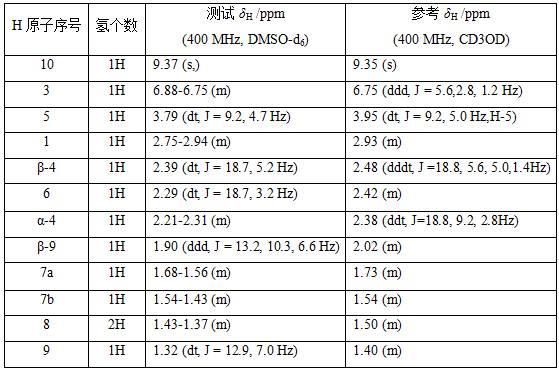
23.图7为单体化合物ⅰ对螯合亚铁离子的能力图。
24.图8为单体化合物ⅰ对fe
3+
的还原能力图。
具体实施方式
25.下面将本发明的发明目的、技术方案和有益效果作进一步详细的说明。
26.应该指出,以下详细说明都是示例性的,旨在对所要求的本发明提供进一步的说明,除非另有说明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
27.刘小玲等在“香辛料草果中化学成分的定性研究”(《中国调味料》,2011年第1期总第36卷,104
‑
106)中通过水提取、乙醇提取、石油醚提取等提取方法对草果中的化学成分进行较全面的定性研究,表明了草果中含有糖类、蛋白质、氨基酸、酚类、鞣质、有机酸、皂苷、黄酮、蒽醌、香豆素、内酯、强心苷、甾体、萜类、挥发油、油脂、花青素等多种化学成分。由此可以知道,草果具有丰富的化学成分,而现有的草果提取物抗氧化活性物质的研究,通常针对草果中某一类提取物而进行,但这些现有研究中并未阐明草果抗氧化活性的物质基础。例如袁园等在“草果总黄酮的提取及dpph自由基清除活性研究”中采用超声波辅助法提取草果总黄酮,并通过试验证明草果总黄酮的dpph自由清除率为80.5%。又如李志君等在“草果多酚物质提取及lc
‑
ms/ms分析”(《食品工业科技》,2017年第08期,294
‑
334)中采用提油后的草果粉末进行多酚物质提取,经试验证明,该草果多酚具有一定的dpph、abts自由基清楚能力,且多酚浓度与其抗氧化活性正相关。
28.因此,有必要设计一种新从草果提取抗氧化活性单体化合物的方法,该方法不仅能够分离得到所需的抗氧化活性物质,还能通过结构鉴定,确定其抗氧化活性的物质基础,为草果进一步的综合开发利用奠定基础。
29.下面以几个典型实施例来列举说明本发明的具体实施方式,当然,本发明的保护范围并不局限于以下实施例。以下实施例中涉及的百分数均为质量份数,使用的仪器、药品与试剂如下:仪器:bc
‑
r501ca型5l旋蒸机组(上海贝凯生物化学有限公司);bc
‑
r2012b型20l旋蒸机组(上海贝凯生物化学有限公司);dlsb
‑
30/130型低温冷却循环泵(郑州金育科贸有限公司);bk
‑
1000b型超声波清洗机(济南巴克超声波科技有限公司);lpg
‑
5型离心喷雾干燥器(四川万泰机械设备有限公司);vfd
‑
1000型vdf冷冻干燥机(北京博医康实验仪器有限公司);biobase bk
‑
fd10p型冷冻干燥机(山东博科生物产业有限公司);hc
‑
5002s型超声仪(昆山慧超自动化设备有限公式);x85
‑
2s型恒温磁力搅拌器(上海梅颖浦仪器仪表制造公司);hh
‑
z4型数显恒温水浴锅(金坛市城东光芒仪器厂);100*200mm硅胶薄层板(青岛海浪硅胶干燥剂有限公司);200
‑
300目硅胶(青岛海浪硅胶干燥剂有限公司);waters2695型制备液相色谱仪(waters);安捷伦1100型分析液相色谱仪(安捷伦);制备液相静态压缩柱50mm(成都市武侯区虹禹药机设备厂);2500ml抽滤瓶(成都振源玻璃仪器有限公司)。
30.药品与试剂:草果药材购自广西玉林市,经鉴定为姜科豆蔻属多年生草本植物草果(a. tsao
‑
ko crevost et lemarie)的干燥果实。将其粉碎成粗粉,于通风干燥条件下贮存,备用。二氯甲烷、乙腈、95%乙醇、甲醇、无水乙醇、磷酸、正丁醇(以上试剂均来自成都市科隆化学品有限公司 ar);2,6
‑
二叔丁基
‑4‑
甲基苯酚(bht,上海晶纯试剂有限公司,优级纯);dpph、abts(上海金穗生物科技有限公司 ar);fecl3·
6h2o(上海实意化学试剂有限公司)。
31.实施例1:从草果中提取分离草果素选取草果药材38kg进行粉碎,95%乙醇回流提取3次,每次2h,55℃减压浓缩至无醇,得到浓缩液后,加水稀释3倍,用等体积的乙酸乙酯萃取2次,将萃取得到的乙酸乙酯提取物在45℃下减压浓缩制得乙酸乙酯提取物浸膏,用其质量2倍的硅胶拌样,拌样物在40℃下鼓风干燥恒重,得到干燥的硅胶拌样物。将硅胶拌样物进行正相硅胶柱层析,以10倍的硅胶为填料,湿法装柱,干法上样,用体积比为30︰1的二氯甲烷
‑
甲醇为洗脱剂进行常压洗脱,分管收集,每管约100ml,再进行薄层色谱分析,以体积比为20︰1的二氯甲烷
‑
甲醇为展开剂,在254 nm的紫外光下检视暗斑结果,将含有目标化合物的收集液合并后,在45℃下减压浓缩至浸膏。再以甲醇
‑
水(50︰50)为流动相,λ=254nm,c18反相色谱填料高压制备分离, 得到目标成分,经45℃浓缩干燥,40℃减压干燥,得到一种单体化合物ⅰ(200mg),得率为0.53%(mg/g)。
32.实施例2:从草果中提取分离草果素选取草果药材 38 kg进行粉碎,95%乙醇回流提取3次,每次1.5h,55℃减压浓缩至无醇,得到浓缩液后,加水稀释2倍,用等体积的乙酸乙酯萃取2次,将萃取得到的乙酸乙酯提取物在45℃下减压浓缩制得乙酸乙酯提取物浸膏,用等质量的硅胶拌样,拌样物在40℃下鼓风干燥恒重,得到干燥的硅胶拌样物。将硅胶拌样物进行正相硅胶柱层析,以20倍的硅胶为填料,湿法装柱,干法上样,用体积比为50︰1的二氯甲烷
‑
甲醇为洗脱剂进行常压洗脱,分管收集,每管约100ml,再进行薄层色谱分析,以体积比为20︰1的二氯甲烷
‑
甲醇为展开剂,在254nm的紫外光下检视暗斑结果,将含有目标化合物的收集液合并后,在45℃下减压浓缩至浸膏。再以甲醇
‑
水(50︰50)为流动相,λ=254nm,c18反相色谱填料高压制备分离, 得到目标成分,经45℃浓缩干燥,40℃减压干燥,得到一种单体化合物ⅱ(210mg),得率为0.55%(mg/g)。
33.实施例3:从草果中提取分离草果素选取草果药材38kg进行粉碎,95%乙醇回流提取2次,每次1.5h,50℃减压浓缩至无醇,得到浓缩液后,加水稀释2倍,用等体积的乙酸乙酯萃取2次,将萃取得到的乙酸乙酯提取物在45℃下减压浓缩制得乙酸乙酯提取物浸膏,用其质量2倍的硅胶拌样,拌样物在40℃下鼓风干燥恒重,得到干燥的硅胶拌样物。将硅胶拌样物进行正相硅胶柱层析,以15倍的硅胶为填料,湿法装柱,干法上样,用体积比为30︰1的二氯甲烷
‑
甲醇为洗脱剂进行常压洗脱,分管收集,每管约100ml,再进行薄层色谱分析,以体积比为20︰1的二氯甲烷
‑
甲醇为展开剂,在254 nm的紫外光下检视暗斑结果,将含有目标化合物的收集液合并后,在45℃下减压浓缩至浸膏。再以甲醇
‑
水(50︰50)为流动相,λ=254nm,c18反相色谱填料高压制备分离, 得到目标成分,经45℃浓缩干燥,40℃减压干燥,得到一种单体化合物ⅲ(198mg),得率为0.52%(mg/g)。
34.实施例4:从草果中提取分离草果素选取草果药材38kg进行粉碎,95%乙醇回流提取3次,每次1.5h,50℃减压浓缩至无醇,得到浓缩液后,加水稀释3倍,用等体积的乙酸乙酯萃取2次,将萃取得到的乙酸乙酯提取物在45℃下减压浓缩制得乙酸乙酯提取物浸膏,用其质量2倍的硅胶拌样,拌样物在40℃下鼓风干燥恒重,得到干燥的硅胶拌样物。将硅胶拌样物进行正相硅胶柱层析,以20倍的硅胶为填料,湿法装柱,干法上样,用体积比为30︰1的二氯甲烷
‑
甲醇为洗脱剂进行常压洗脱,
分管收集,每管约100ml,再进行薄层色谱分析,以体积比为20︰1的二氯甲烷
‑
甲醇为展开剂,在254nm的紫外光下检视暗斑结果,将含有目标化合物的收集液合并后,在45℃下减压浓缩至浸膏。再以甲醇
‑
水(50︰50)为流动相,λ=254nm,c18反相色谱填料高压制备分离, 得到目标成分,经45℃浓缩干燥,40℃减压干燥,得到一种单体化合物ⅳ(202mg),得率为0.53%(mg/g)。
35.实施例5:从草果中提取分离草果素选取草果药材38kg进行粉碎,95%乙醇回流提取2次,每次2h,55℃减压浓缩至无醇,得到浓缩液后,加水稀释2倍,用等体积的乙酸乙酯萃取2次,将萃取得到的乙酸乙酯提取物在45℃下减压浓缩制得乙酸乙酯提取物浸膏,用等质量的硅胶拌样,拌样物在40℃下鼓风干燥恒重,得到干燥的硅胶拌样物。将硅胶拌样物进行正相硅胶柱层析,以15倍的硅胶为填料,湿法装柱,干法上样,用体积比为30︰1的二氯甲烷
‑
甲醇为洗脱剂进行常压洗脱,分管收集,每管约100ml,再进行薄层色谱分析,以体积比为20︰1的二氯甲烷
‑
甲醇为展开剂,在254 nm的紫外光下检视暗斑结果,将含有目标化合物的收集液合并后,在45℃下减压浓缩至浸膏。再以甲醇
‑
水(50︰50)为流动相,λ=254nm,c18反相色谱填料高压制备分离, 得到目标成分,经45℃浓缩干燥,40℃减压干燥,得到一种单体化合物
ⅴ
(201mg),得率为0.53%(mg/g)。
36.实施例6:单体化合物ⅰ的结构鉴定将上述所得的单体化合物ⅰ进行紫外光谱(uv)、核磁共振(1h
‑
nmr 和 13
c
‑
nmr)、质谱(ms)等测试。
37.(1)紫外光谱单体化合物的紫外吸收曲线在波长为234.3nm处出现最大吸收峰,说明可能含有两个共轭双键,在波长为306.8nm处出现低强度的吸收峰,(n
→
π*跃迁),说明可能含有羰基。参见图1。
38.(2)核磁共振氢谱(1h nmr):此化合物的核磁共振氢谱信号归属为:1h nmr(dmso
‑
d6, 400 mhz)δ: 9.37(1h, s, h
‑
10),6.88
‑ꢀ
6.75(1h, m, h
‑
3),3.79(1h, dt, j=9.2, 4.7hz, h
‑
5),2.75
‑
2.94(1h, m, h
‑
1),2.39(1h, dt, j=18.7, 5.2hz, h
β
ꢀ‑
4),2.29(1h, dt, j=18.7, 3.2hz h
‑
6),2.21
‑
2.31(1h, m, h
α
ꢀ‑
4),1.90(1h, ddd, j=13.2, 10.3, 6.6hz, h
β
ꢀ‑
9),1.68
‑
1.56(1h, m, h
‑
7a),1.54
‑
1.43(1h, m, h
‑
7b),1.43
‑
1.37(2h, m, h
‑
8),1.32(1h, dt, j=12.9, 7.0hz, h
ꢀ‑
9)。见下表1。
39.表1 核磁共振氢谱的信号归属(1h nmr)
(3)核磁共振碳谱(
13
c nmr):此化合物的核磁共振碳谱信号归属为:
13
c nmr(dmso
‑
d6, 100 mhz)δ:37.06(c
‑
1),144.29(c
‑
2),149.22(c
‑
3),31.5(c
‑
4),66.71(c
‑
5),42.82(c
‑
6),25.28(c
‑
7),24.7(c
‑
8),32.2(c
‑
9),194.67(c
‑
10)共出现10个碳信号。见下表2。
40.表2核磁共振碳谱的信号归属(
13
c nmr)(4)质谱:质谱esi+中给出的准分子离子峰[2m+li]
+
:m/z 339.34。说明该化合物分子量m=166.22。参见图2。
[0041]
(4)结构式:
综上,根据核磁共振(1h nmr、
13
c nmr)与质谱结果,再与现有文献(王剑丽等“花生衣化学成分的分离与结构鉴定”,《天津中医药大学学报》,2019,38(2):175
‑
179)对照,确定其结构为原儿茶酸(3,4
‑
二羟基苯甲酸),属于酚酸类化合物。白色粉末,分子式为c7h6o4,分子量为154.12。
[0042]
综上,根据紫外光谱,核磁共振(1h nmr、
13
c nmr)与质谱结果,再与现有文献(金家宏等“四种药用植物的抗补体活性成分”,《复旦大学》, 2011:103
‑
114)对照,确定其结构为草果素(2
‑
(丁烯基)
‑4‑
羟基
‑3‑
甲基
‑2‑
环戊烯
‑1‑
酮),属于萜类化合物。淡黄色液体,分子式为c
10
h
12
o2,分子量为166.22。具体结构式如下式(1)。
[0043]
ꢀꢀ
(1)实施例7:单体化合物ⅰ的体外抗氧化活性测定取单体化合物ⅰ及对照品bht,制备成浓度为:5、10、20、30、40 μg/ml 的系列工作溶液,于4℃低温保存,备用。
[0044]
(1)dpph自由基清除能力的测定取单体化合物ⅰ以及bht工作溶液分别进行dpph自由基的清除试验。单体化合物及bht均设置11个具塞比色管,编为0~10号:在0号比色管中加入0.1 mmol/l dpph工作溶液2 ml和无水乙醇2 ml,作为空白管;在1~5号比色管中分别加入0.1 mmol/l dpph工作溶液2 ml和相应药物的系列工作液(5、10、20、30、40 μg/ml)各2 ml,作为试验管;在6~10号管中分别加入无水乙醇2 ml和相应药物的系列工作液(5、10、20、30、40 μg/ml)各2 ml,作为对照管。各比色管混匀后,在30℃水浴下避光反应30 min。以无水乙醇进行调零,采用紫外
‑
可见分光光度计在517 nm波长处测定各反应溶液的吸光度(a)值,按公式计算dpph自由基清除率:dpph自由基清除率(%)=[1-(a试验-a对照)/a 空白]
×
100%。
[0045]
a空白: 2 ml dpph工作液+ 2 ml 无水乙醇;a试验: 2 ml dpph工作液+2 ml 相应药物的系列工作液;a对照: 2ml无水乙醇+2 ml 相应药物的系列工作液。
[0046]
测定结果如图3所示,随着样品浓度的增大,对照品bht以及草果素对dpph自由基的清除能力逐渐增大,对照品bht对dpph自由基的清除能力明显比草果素强。
[0047]
(2)abts自由基清除能力的测定取单体化合物ⅰ以及bht工作溶液分别进行超氧阴离子自由基清除试验。单体化合物及bht均设置11个具塞比色管,编为0~10号:0号比色管加入6 ml abts工作液和0.6 ml 无水乙醇作为空白管, 1~5号比色管加入6 ml abts工作液和相应药物的系列工作液(5、10、20、30、40 μg/ml)各0.6 ml,作为试验管;6~10号比色管加入6 ml 无水乙醇和相应药物的系列工作液(5、10、20、30、40 μg/ml)各 0.6 ml,作为对照管。各比色管混匀后,室温避光反应1h,以无水乙醇进行调零,采用紫外
‑
可见分光光度计在734 nm波长处测定各反应溶液的吸光度(a)值,按公式计算abts自由基清除率:abts自由基清除率(%)=[1-(a试验-
a对照)/a空白]
×
100%。
[0048]
a空白:6 ml abts 工作液+0.6 ml 无水乙醇;a试验:6 ml abts 工作液+0.6 ml相应药物的系列工作液;a对照:6 ml无水乙醇+0.6 ml相应药物的系列工作液。
[0049]
测定结果如图4所示,随着样品浓度的增大,草果素对abts自由基的清除能力并不明显,增长趋势呈现持平现象,而对照品bht对abts自由基的清除能力较强。
[0050]
(3)超氧阴离子自由基清除能力的测定取单体化合物ⅰ以及bht工作溶液分别进行超氧阴离子自由基清除试验。单体化合物及bht均设置11个具塞比色管,编为0~10号:0号比色管加入1 mol/l tris
‑
hcl缓冲液(ph 7.4)4 ml、2.5 mmol/l邻苯三酚溶液0.4 ml和无水乙醇1 ml,作为空白管;1~5号比色管加入1 mol/l tris
‑
hcl缓冲液4 ml、2.5mmol/l邻苯三酚溶液0.4 ml和相应药物的系列工作液(5、10、20、30、40 μg/ml)各1 ml,作为试验管;6~10号比色管加入1 mol/l tris
‑
hcl 缓冲液4 ml、蒸馏水0.4 ml和相应药物的系列工作液(5、10、20、30、40 μg/ml)各1 ml,作为对照管。各比色管混匀后,在室温下反应5 min,然后加入8 mmol/l hcl溶液1 ml终止反应。以蒸馏水进行调零,采用紫外
‑
可见分光光度计在320 nm波长处测定各反应溶液的a值,按公式计算超氧阴离子清除率:超氧阴离子清除率(%)=[1-(a试验-a对照)/a空白]
×
100%。
[0051]
a空白:4 ml tris
‑
hcl+ 1 ml无水乙醇+ 0.4 ml邻苯三酚+1 ml 8 mmol/l hcl的吸光度值;a试验:4 ml tris
‑
hcl + 1 ml 相应药物的系列工作液+ 0.4 ml 邻苯三酚+1 ml 8 mmol/l hcl的吸光度值;a对照:4 ml tris
‑
hcl + 1 ml 相应药物的系列工作液+ 0.4 ml 蒸馏水+1 ml 8 mmol/l hcl的吸光度值。
[0052]
测定结果如图5所示,在设定的浓度范围内,随浓度的变化,草果素及bht对超氧阴离子的清除能力变化不太明显,但草果素对超氧阴离子阴离子自由基的清除能力明显比对照品bht强。
[0053]
(4)羟基自由基清除能力的测定取单体化合物ⅰ以及bht工作溶液分别进行羟基自由基清除试验。单体化合物及bht均设置6个具塞比色管,编为0~5号:0号比色管加入1 ml feso4溶液、1 ml水杨酸-乙醇溶液、1 ml蒸馏水、1 ml h2o2溶液,作为空白管,1~5号比色管加入1 ml feso4溶液、1 ml水杨酸-乙醇溶液、1 ml h2o2溶液和相应药物的系列工作液(5、10、20、30、40 μg/ml)各1 ml,作为试验管,以蒸馏水进行调零,采用紫外
‑
可见分光光度计在510 nm波长处测定各反应溶液的a值,按公式计算羟基自由基清除率:羟基自由基清除率(%)=[a空白-a试验/a空白]
×
100%。
[0054]
a空白:1 ml feso4溶液+1 ml水杨酸-乙醇溶液+1 ml蒸馏水+1 ml h2o2溶液;a试验:1 ml feso4溶液+1 ml水杨酸-乙醇溶液+1 ml h2o2溶液+1 ml相应药物的系列工作液。
[0055]
测定结果如图6所示,草果素和对照品bht对羟基自由基具有一定的清除能力,且其清除能力随浓度的增大而增大,但对照品bht对羟基自由基清除能力比较强。
[0056]
(5)与金属离子(fe
2+
)螯合能力的测定取单体化合物ⅰ以及bht工作溶液分别进行fe
2+
螯合能力试验。单体化合物及bht均设置6个具塞比色管,编为0~5号:0号比色管加入0.1 ml fecl2、0.2 ml 5 mmol/l菲啰嗪、5ml蒸馏水,作为空白管,1~5号比色管加入0.1 ml fecl2、0.2 ml 5 mmol/l菲啰嗪、3 ml蒸馏水和相应药物的系列工作液(5、10、20、30、40μg/ml)各2 ml,作为试验管,以蒸馏水进行调零,反应10 min后,采用紫外
‑
可见分光光度计在562 nm波长处测定各反应溶液的a值,吸光度越低表示金属螯合能力越高。按公式计算亚铁离子螯合能力:亚铁离子螯合能力(%)=[a空白-a试验/a空白]
×
100%。
[0057]
a空白:5 ml蒸馏水+0.1 ml fecl2+0.2 ml 5 mmol/l菲啰嗪;a试验:3 ml蒸馏水+0.1 ml fecl2+0.2 ml 5 mmol/l菲啰嗪+2 ml相应药物的系列工作液。
[0058]
测定结果如图7所示,草果素与对照品bht在浓度为5
µ
g/ml时,其对fe
2+
螯合能力接近相等,但随着浓度的增大,草果素对fe
2+
螯合能力的变化最大。
[0059]
(6) fe
3+
还原能力的测定取单体化合物ⅰ以及bht工作溶液分别进行fe
3+
还原试验。单体化合物及bht均设置15 ml离心管5支,分别加入相应药物的系列工作溶液(5、10、20、30、40 μg/ml)各1 ml、0.2 mol/l的磷酸盐缓冲液(ph 6.6)2.5 ml和1%铁氰化钾溶液2.5 ml。各离心管在50℃水浴中加热反应20 min后,加入10%三氯乙酸溶液2.5 ml终止反应,然后以3000 r/min离心10 min;取上清液2.5 ml于具塞比色管中,加入蒸馏水2.5 ml 和0.1%fecl3溶液0.5 ml,混匀,静置反应10 min。以蒸馏水进行调零,采用紫外
‑
可见分光光度计在波长700 nm处测定各反应溶液的a值。以a值表示待测药物对fe
3+
的还原能力,a值越大,则表示药物的还原能力越强。
[0060]
a样品:[1 ml 样品+2.5 ml ph 6.6 的磷酸盐缓冲液+2.5 ml 1%的铁氰化钾溶液]水浴20 min+三氯乙酸溶液 2.5 ml终止反应(离心) 取上清2.5 ml+2.5 ml 蒸馏水+0.5 ml 0. 1%的fecl3溶液a对照:[1 ml bht+2.5 ml ph 6.6 的磷酸盐缓冲液+2.5 ml 1%的铁氰化钾溶液] 水浴20 min+三氯乙酸溶液2.5 ml终止反应(离心) 取上清2.5 ml+2.5 ml 蒸馏水+0.5 ml 0.1%的fecl3溶液。
[0061]
测定结果参见图8。吸光度值越大,说明还原能力越大,即还原能力越强,说明抗氧化能力越强,由图8所示,随着浓度的增大,对照品bht和草果素的吸光度值也增大,但照品bht的吸光度值变化最明显。
[0062]
基于上述实施例,本发明提供了一种提取草分离草果中单体化合物草果素的方法。草果素属于萜类化合物,萜类化合物是指以异戊二烯为骨架的烃类及其含氧衍生物,结构中的烯键化学性质活泼,具有较强的还原性(即抗氧化性),能跟氧化剂起反应,从而保护机体不受氧化损伤
[1]
。单体化合物对不同的自由基清除能力不同,可能是与化合物的结构特征有关,包括:空间结构、羟基的位置、羟基的数量、苯环上的酚羟基等
[2]
。
[0063]
通过dpph自由基清除实验、abts自由基清除实验、超氧阴离子自由基清除实验、羟基自由基清除实验、亚铁离子的螯合实验、三价铁离子的还原实验发现,草果素对这些自由基及铁离子均具有一定的清除能力,说明草果素具有一定的抗氧化能力,其抗氧化活性与
单体化合物结构有关,抗氧化作用机理主要是通过清除自由基来避免氧化损伤。
[0064]
[1] 贺娟, 梁惠, 史大永, 等. 凹顶藻提取物的抗氧化作用[j]. 中国公共卫生, 2005, 21(9):1082
‑
1083.[2] 赵雪巍, 刘培玉, 刘丹, 等. 黄酮类化合物的构效关系研究进展[j]. 中草药, 2015, 46(21):3264
‑
3271.实施例8:对上述实施例1
‑
5中从草果中提取分离草果素的方法进行考察,其方法学验证过程如下:(1)精密度试验:精密吸取草果素溶液 10
µ
l 进样1次,记录色谱图。结果:峰面积的rsd值为0.13%,说明仪器精密度良好。
[0065]
(2)稳定性试验:精密吸取草果素溶液,分别于0、2、4、8、12、24h进样,测定峰面积。结果:草果素峰面积的rsd值为0.43%,说明草果素溶液在24h内稳定。
[0066]
(3)重复性试验:取草果素样品6份,每份约1mg,精密称定,按供试品溶液的制备方法制备供试品溶液,按色谱条件进行测定。结果:草果素含量为0.53%(mg/g),rsd值为0.83%,说明方法重复性良好。
[0067]
(4)加样回收率试验:取已测定的草果素约0.5mg,精密称定,平行称取5份,置具塞锥形瓶中,按标准品与供试品含量比为0.5︰1、1︰1、1.5︰1分别加入标准品,每个比例配置3份,制备供试品溶液。各供试品溶液进样体积10μl,测定峰面积,计算回收率。结果:草果素平均回收率为100.18%,rsd为1.37%,结果表明方法回收率良好。
[0068]
以上所述,仅是本发明的较佳实施例,并非对本发明做任何形式上的限制,凡是依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化,均落入本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1