一种含硒抗流感药物的制备方法与流程
1.本发明涉及生物医药领域,具体涉及一种含硒抗流感药物的制备方法。
背景技术:
2.流感病毒属于正粘病毒科(orthomyxoviridae),其是含有单链负义rna基因组的包膜病毒。在过去几十年里,通常两类疗法可用于治疗流感病毒:m2离子通道抑制剂和神经氨酸酶抑制剂。然而,流感病毒不仅表现出对m2离子通道抑制剂的广泛抗药性,而且陆续产生对神经氨酸酶抑制剂的抗病毒抗药性。
3.cap(帽)依赖性核酸内切酶抑制剂通过控制流感病毒复制的关键环节,抑制病毒从宿主细胞中获得宿主mrna 5’端的cap结构,从而抑制流感病毒自身mrna的转录,达到治疗流感的效果。而且由于宿主细胞内不存在有类似机制的蛋白酶,cap依赖性核酸内切酶抑制剂不会对宿主细胞产生影响。2018年,首个帽依赖性内切核酸酶(cen)抑制剂巴洛沙韦酯(baloxavir marboxil)(商品名:xofluza))在美国和日本批准用于治疗a型和 b型流感。2021年4月29日,巴洛沙韦酯获得nmpa批准在中国上市,适应症为治疗12 周岁及以上的流感患者,包括存在流感并发症高风险的患者。前药巴洛沙韦酯在体内代谢转化为活性成分巴洛沙韦。巴洛沙韦抑制流感病毒聚合酶酸性(pa)蛋白质内切核酸酶,使病毒rna的合成得以抑制,从而可以有效抑制流感病毒复制。
4.盐野义公司专利cn109311911a中公开了一种巴洛沙韦的制备方法,其反应路线如下:
[0005][0006]
中间体13通过转换取代基(产率为87.2%)、偶联(产率为84.6%)和脱保护(产率为90.7%)等工序后得到式v所示的巴洛沙韦。上述过程不仅反应路线较长,操作繁琐,且从中间体13得到式v所示的巴洛沙韦的总收率仅为65%左右。
[0007]
公开号为wo2021007506a1的专利申请中提供了一种新型的含硒抗流感药物,其作为一种cap依赖性核酸内切酶抑制剂,能够用来治疗流感,显示出了优异的生物活性及药代动力学性质包括良好的口服生物利用度,且不受进食的影响。当采用和cn109311911a中类
似的方法,来制备所述的含硒抗流感药物时,反应路线如下:
[0008][0009]
中间体11-r通过转换取代基(产率为78.2%)、偶联(产率为32.6%)和脱保护(产率为86.3%)等工序后得到式b-1所示的含硒抗流感药物。从中间体11-r出发,经过三步反应得到b-1所示的含硒抗流感药物,总收率仅为22%。
[0010]
终上所述,针对含硒的抗流感药物,需要开发新的制备方法。
技术实现要素:
[0011]
针对现有技术的不足,本发明的目的是提供一种含硒抗流感药物的制备方法,该方法工艺路线简单、选择性好、总收率高、适宜工业化生产。具体而言,该方法不需要经过转换取代基和脱保护基工序,只通过一步偶联工序就可以选择性地生成含硒抗流感药物,在简化反应路线的同时大大提升了收率,十分适宜工业化生产。
[0012]
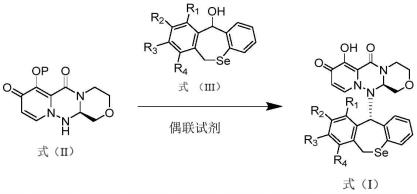
本发明所述的含硒抗流感药物如式(i)所示,式(i)化合物可以用来治疗流感或者制备其他治疗流感的药物,其结构如下所示:
[0013][0014]
其中,r1、r2、r3和r4各自独立地选自氢或者卤素;优先地,r1、r2、r3和r4中的一个或者两个为卤素。
[0015]
一种式(i)所示化合物的制备方法,所述合成方法包括:式(ii)所示的吡啶并三嗪二酮衍生物与式(iii)所示的二氢二苯并硒平衍生物在偶联试剂的存在下,生成式(i)化合物,其反应路线如下所示:
[0016][0017]
其中,其中,r1、r2、r3和r4各自独立地选自氢或者卤素;优先地,r1、r2、r3和 r4中的一个或者两个为卤素;更优选地,r1和r2为氢,r3和r4为氟;
[0018]
p为羟基保护基;优选地,所述羟基保护基为苄基(bn)、取代苄基、苄氧羰基(cbz)、三苯基甲基、叔丁基二甲基硅基(tbdms)、三甲基硅基(tms)、叔丁基二苯基硅基(tbdps)、三乙基硅基(tes)、三异丙基硅基(dips)、2-(三甲硅烷基)乙氧甲基、二氢吡喃基、溴丙烯基、乙酯甲酰基、乙酰基或苯甲酰基等;更优选地,所述羟基保护基为苄基(bn)、或在苄基的苯环上被1-2个甲基,甲氧基,或卤素取代的苄基。;
[0019]
偶联试剂为磷酸酐试剂,其中磷酸酐试剂包括烷基磷酸酐和芳基磷酸酐,其中烷基磷酸酐如:丙基磷酸酐(t3p),乙基甲基次膦酸酐(empa);芳基磷酸酐,如:苯基膦酸酐;偶联试剂也可以为其他磷酸酯和磷酰胺类缩合剂,如:二苯基磷酰氯(dpp-cl)、氰代磷酸二乙酯(decp)、叠氮化磷酸二苯酯(dppa)、硫代二甲基磷酰基叠氮(mpta)、二(2-氧-3-唑烷基)磷酰氯(bop-cl)。优选地,所述磷酸酐试剂为烷基磷酸酐试剂,更优选地为丙基磷酸酐(t3p)。
[0020]
在一些实施例中,本发明的制备方法,进一步包括如下步骤:
[0021]
步骤1:室温(15~35℃)下,将式(ii)化合物和式(iii)溶解于有机溶剂1中,将反应液冷却至-10℃~10℃后,滴加酸性试剂1,滴加完毕后,将反应液加热至室温,直至反应液由澄清变为浑浊;
[0022]
步骤2:向步骤1得到的浑浊溶液中滴加偶联试剂溶液和酸性试剂2,升温到40℃至 100℃,继续反应12-48小时;
[0023]
步骤3:监测反应完成后,进行后处理,得到式(i)化合物。
[0024]
可选的实施方案中,步骤1中所述的有机溶剂1选自低级烷烃类溶剂、卤代低级烷烃类溶剂、醚类溶剂、腈类溶剂、酮类溶剂、酯类溶剂、酰胺类溶剂或亚砜类溶剂中的一种或多种;优选地,选自酯类溶剂;更优选地,酯类溶剂选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、乙酸异丁酯、乙酸正戊酯、乙酸异戊酯和乙酸新戊酯中的一种或多种。
[0025]
可选的实施方案中,步骤1中所述的酸性试剂1和步骤2中所述的酸性试剂2相同或不同,为无机酸或有机酸;优选地,所述无机酸选自盐酸、硫酸、硝酸、碳酸、氢溴酸、磷酸等;优选地,所述有机酸为甲酸、乙酸、丙酸、三氟乙酸、柠檬酸、乳酸、酒石酸、草酸、马来酸、富马酸、戊二酸、苹果酸、苯甲酸、苯二甲酸、抗坏血酸、苯磺酸、对甲苯磺酸、甲磺酸、三氟甲磺酸、乙磺酸等;更优选地,所述的酸性试剂1和酸性试剂2相同或不同,分别选自苯磺酸、对甲苯磺酸、甲磺酸、三氟甲磺酸和乙磺酸中的一种或多种。最优选地,酸性试剂1为三氟甲磺酸,酸性试剂2为甲磺酸。
[0026]
可选的实施方案中,所述偶联试剂溶液,由丙基磷酸酸酐(t3p)溶于酯类溶剂中产
生,所述酯类溶剂选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、乙酸异丁酯、乙酸正戊酯、乙酸异戊酯和乙酸新戊酯中的一种或多种。
[0027]
可选的实施方案中,所述偶联试剂本身为液体状态的,也可以不溶于其他溶剂,直接滴加到反应体系当中。
[0028]
可选的实施方案中,式(ii)化合物与式(iii)化合物的摩尔当量比为1:1至1:1.5,更优选地为1:1~1:1.2。
[0029]
可选的实施方案中,式(ii)化合物与酸性试剂1的摩尔当量比为1:1至1:2,更优选地为1:1~1:1.5。
[0030]
可选的实施方案中,式(ii)化合物与酸性试剂2的摩尔当量比为1:1至1:5,更优选地为1:2~1:5。
[0031]
本发明的有益效果
[0032]
1.工艺路线简短(一步法),避免保护基的重复保护与脱去,条件温和,总收率高;十克级别工艺稳定,适宜工业化生产。
[0033]
2.本发明工艺选择高,可以有效减少式(i)化合物对映异构体的产生,所述式(i)化合物对映异构体的结构如式(i-a)所示,反应完成时,反应液中式(i-a)含量低于5%, r1、r2、r3和r4的定义如式(i)中所述。
[0034]
附图说明
[0035]
图1是本发明的实施例1反应完成时反应液的hplc检测结果图。
[0036]
图2是本发明的实施例1终产物的hplc检测结果图。
[0037]
图3是本发明的实施例2反应完成时反应液的hplc检测结果图。
[0038]
图4是本发明的实施例2终产物的hplc检测结果图。
具体实施方式
[0039]
下面的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。同时,对于文中出现的一些术语进行解释和说明,这些解释和说明仅用于方便本领域技术人员理解,不应看作是对本发明保护范围的限制。
[0040]
所提到的“卤素”指氟,氯,溴或碘。
[0041]
所提到的“羟基保护基团”是指羟基的取代基用来阻断或保护羟基的功能性,合适的保护基团包括但不限于苄基(bn)、取代苄基、苄氧羰基(cbz)、三苯基甲基、叔丁基二甲基硅基(tbdms)、三甲基硅基(tms)、叔丁基二苯基硅基(tbdps)、三乙基硅基(tes)、三异丙基
硅基(dips)、2-(三甲硅烷基)乙氧甲基、二氢吡喃基、溴丙烯基、乙酯甲酰基、乙酰基或苯甲酰基等。
[0042]
所提到的“取代苄基”或“取代的苄基”是指苄基的苯环上被一个或多个甲基,甲氧基,或卤素取代;如苄基的苯环上被1-2个甲基,甲氧基,或卤素取代的苄基;包括但不限于对甲氧基苄基和2,4-二甲氧基苄基。
[0043]
所提到的“有机溶剂”可以选自低级烷烃类溶剂、卤代低级烷烃类溶剂、醚类溶剂、腈类溶剂、酮类溶剂、酯类溶剂、酰胺类溶剂或亚砜类溶剂中的一种或多种;所述的低级烷烃类溶剂可以选自环己烷、正己烷、正庚烷、正戊烷、异戊烷、石油醚、异辛烷或环戊烷等;所述的卤代低级烷烃类溶剂可以选自二氯甲烷或氯仿;所述的醚类溶剂可以选自四氢呋喃、二氧六环、异丙醚或甲基叔丁基醚;所述的腈类溶剂可以选自乙腈或丙腈;所述的酮类溶剂可以选自丙酮、甲乙酮或丁酮;所述的酯类溶剂可以选自乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸正丁酯、乙酸异丁酯、乙酸正戊酯、乙酸异戊酯或乙酸新戊酯;所述酰胺类溶剂可以选自n,n-二甲基甲酰胺(dmf)或n-甲基吡咯烷酮(nmp);所述亚砜类溶剂选自二甲亚砜(dmso)。
[0044]
在描述hplc的结果时,虽然对于保留时间给出了具体的数值。但是本领域技术人员可知的是,这些保留时间的具体数值仅作参考,实际在进行检测时,保留时间数值可能会上下波动。在用“约”表示保留时间时,就反映了这种保留时间的变化。
[0045]
实施例1
[0046][0047]
室温下,将式(ii)化合物(20.0g,61mmol)和式(iii)化合物(21.1g,67mmol) 溶解于140ml乙酸正丁酯(buoac)中,待完全溶解后,将反应体系降温至0~10℃,缓慢滴加三氟甲磺酸(tfoh,9.2g,61mmol),滴加完成后升温至室温,得到悬浮液。继续向反应液中滴加重量比为50%的丙基磷酸酐(t3p)乙酸乙酯溶液38.9g,其中t3p含量为 19.45g(61mmol);并滴加甲磺酸(17.6g,183mmol),滴加完成后,将反应体系升温至60℃,继续反应。在反应进行到第23小时,进行hplc监测,其中式(ⅱ)化合物含量为4.99%,式(i)化合物含量为76.62%,式(i-a)的含量为3.23%,具体分析结果见附图1。反应结束后,冷却至室温。抽滤后得滤饼,buoac洗涤后,将滤饼溶于4倍体积的乙腈中,而后滴加甲基叔丁基醚(mtbe)至固体全部析出,然后经过滤、mtbe和水洗涤、干燥等过程得到式(i)化合物,产率为64.9%,纯度98.56%,其中式(i-a)含量为0.42%。具体分析结果见附图2.
[0048]
式(i)结构鉴定:1h nmr(300mhz,dmso)δ11.80(s,1h),7.45
–
7.28(m,2h),7.24 (d,j=7.0hz,1h),7.17(d,j=7.7hz,1h),7.14
–
7.02(m,2h),6.90(dd,j=10.7,4.1hz,1h), 5.80(s,1h),5.55(d,j=7.7hz,1h),5.29(dd,j=12.7,2.2hz,1h),4.59(dd,j=9.9,2.9hz, 1h),4.43(d,j=12.1hz,1h),4.19
–
3.94(m,2h),3.66(t,j=10.6hz,2h),3.43(dd,j=11.6, 9.3hz,1h),3.15
–
2.97(m,1h).
[0049]
ms实验值532([m+h]
+
)。
[0050]
实施例2
[0051][0052]
室温下,将式(ii)化合物(2.0kg,1.0eq)和式(iii)化合物(2.1kg,1.1eq)溶解于14l乙酸正丁酯(buoac)中,待完全溶解后,将反应体系降温至0~10℃,缓慢滴加三氟甲磺酸(tfoh,918g,1.0eq),滴加完成后升温至室温,得到悬浮液。继续向反应液中滴加重量比为50%的丙基磷酸酐(t3p)乙酸乙酯溶液97.4g,其中t3p含量为3895g(1.0 eq);并滴加甲磺酸(1764g,3.0eq),滴加完成后,将反应体系升温至55-60℃,继续反应。在反应进行到第38小时,进行hplc监测,其中式(ⅱ)化合物含量为4.25%,式(i) 化合物含量为78.80%,式(i-a)的含量为3.35%,具体分析结果见附图3。反应结束后,采用和实施例1类似的方法进行后处理,得到式(i)化合物,产率为66.0%,纯度99.68%,其中式(i-a)含量为0.27%。具体分析结果见附图4。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1