一种双乙氧乙膦烷的制备方法及反应装置与流程
1.本发明涉及化学药品制备的技术领域,具体涉及一种双乙氧乙膦烷的制备方法及反应装置。
背景技术:
2.注射用亚锡替曲膦是一种锝标记药盒,用放射性同位素锝标记得到的
99m
tc-替曲膦(
99m
tc-双乙氧乙膦烷)注射液是一种放射性诊断药物,临床用于心肌灌注显像。
9m
tc-替曲膦注射液的放射化学纯度是影响临床显像质量的关键,而其配体化合物双乙氧乙膦烷(化学名1,2-双[双(2-乙氧基乙基)膦基]乙烷)的纯度是影响其放射化学纯度的关键。
[0003]
双乙氧乙膦烷的合成通常方法通常为:将乙二膦(h2pc2h4ph2化学名1,2-双(膦基)乙烷)和乙烯基乙醚混合,然后加入偶氮二异丁腈,混匀后充氮气密闭,接着置于75℃的油浴反应16小时,后冷却至室温。冷却后将反应物在80℃减压蒸馏1h,在此过程中,未反应完的乙烯基乙醚和乙二膦被除去,偶氮二异丁腈分解成低分子物质不断被蒸走,剩下的即为双乙氧乙膦烷。
[0004]
上述的反应一般在普通玻璃管中进行,使用聚四氟乙烯材质的旋盖旋紧于玻璃管上进行密闭,获得的双乙氧乙膦烷的收率为70%,纯度为98%。目前双乙氧乙膦烷制备完成后还没有方法对其进行纯化,因此想要控制双乙氧乙膦烷的制备控制反应过程很重要,但通过试验发现仅仅通过参数调整并不能有效进一步提高产率和纯度,且现有技术中并没有公开任何可以有效控制反应过程进而将双乙氧乙膦烷的产率和纯度明显提高的记载。
技术实现要素:
[0005]
本发明要解决的技术问题在于克服现有技术中双乙氧乙膦烷的产率和纯度难以提高的缺陷。
[0006]
为此,本发明提供一种双乙氧乙膦烷的制备方法,将反应物放入反应管中后,将反应管端部熔封后再进行油浴反应。
[0007]
可选的,在所有反应物加入完成后,立即向反应管内充入保护气,然后进行熔封。
[0008]
可选的,所述保护气为氮气或惰性气体。
[0009]
可选的,在冰浴条件下添加反应物。
[0010]
可选的,熔封时,将反应管从冰浴条件下取出。
[0011]
可选的,所述反应物包括乙二膦、乙烯基乙醚和偶氮二异丁腈。
[0012]
可选的,添加反应物时,首先向反应管内加入偶氮二异丁腈,接着向反应管内通入保护气再加入其他反应物。
[0013]
可选的,添加反应物时,通过保护气将乙二膦压入反应管中。
[0014]
本发明还提供一种反应装置,其为前述的一种双乙氧乙膦烷的制备方法中所用的反应管,所述反应管包括适于放置反应物的容纳部和适于熔封的熔封部。
[0015]
可选的,所述熔封部的长度大于所述容纳部的长度。
[0016]
可选的,所述反应管为一体成型的玻璃管。
[0017]
本发明技术方案,具有如下优点:
[0018]
1.本发明提供的一种双乙氧乙膦烷的制备方法,发明人在研究发现,在75℃下的油浴化学反应这一步骤中,由于乙烯基乙醚的沸点(35.6℃)较低,在75℃下反应时极易泄露,且油浴化学反应过程中,存在反应压力大、反应时间长、密封效果较差的问题,因此,难以抑制乙烯基乙醚的泄露,进而导致反应物比例失衡,致使乙二膦没有完全反应形成聚合物,最终难以去除,导致产物的收率和纯度下降。基于上述问题的发现,本发明采用将反应物熔封于反应管中后再进行油浴反应的手段,不通过旋盖密封反应管,而是通过熔封实现反应管的密封,大大提高了反应管的密封效果,能够防止反应管中的乙烯基乙醚在油浴过程中挥发泄露,使其能够充分反应,从而有效提高制备的双乙氧乙膦烷的收率和纯度。
[0019]
2.本发明提供的一种双乙氧乙膦烷的制备方法,反应物中的乙二膦化学性质极其活泼,在常温下容易被氧化、空气中容易自燃,因此一般需要收集至充满保护气的集液瓶中,但是在加入反应管的过程中,乙二膦容易暴露在空气中导致其自燃或氧化,而在添加乙二膦前后均向反应管中充入保护气,并通过保护气将乙二膦压入反应管中,能够充分隔绝乙二膦和空气的接触,减少乙二膦的氧化损失,从而降低乙二膦自身化学性质活泼对产物收率和纯度的影响。
[0020]
3.本发明提供的一种双乙氧乙膦烷的制备方法,反应物中的乙二膦在常温下容易被氧化,使反应物比例失衡,影响产物收率和纯度,而在冰浴条件下添加反应物,能够降低反应物的温度,从而降低乙二膦的氧化速率,提高反应物收率和纯度。
[0021]
4.本发明提供的一种双乙氧乙膦烷的制备方法,添加反应物结束后,将反应管从冰浴条件中取出再进行熔封,使熔封过程中反应物温度升高,低沸点和保护气向反应管管口处挥发,防止空气倒灌入反应管中而导致乙二膦氧化,有益于乙二膦的充分反应,保证产物的收率和纯度。
[0022]
5.本发明提供的一种双乙氧乙膦烷的制备方法,在现有的制备双乙氧乙膦烷的方法中,一般通过在手套箱内反应来避免乙二膦的氧化和自燃,以提高产物收率和纯度,而本发明无需在手套箱中进行反应料的添加,即可保证产物的收率和纯度,使双乙氧乙膦烷的生产环境不再苛刻,降低了制备双乙氧乙膦烷的成本。
[0023]
6.本发明提供的一种反应装置,容纳部的长度大于所述反应管的长度,使熔封时热量尽可能小的影响反应物,减小乙烯基乙醚的挥发,保证反应物充分反应。
[0024]
7.本发明提供的一种反应装置,反应管为一体成型的玻璃管,材质完全相同,不会因为材质不同造成油浴过程中出现膨胀系数不同导致的间隙,熔封后密封性较高。
附图说明
[0025]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0026]
图1是本发明实施例1所提供的一种反应装置的示意图;
[0027]
图2是本发明中实施例所提供的收集瓶和反应管的连接示意图。
[0028]
附图标记:1、容纳部;2、熔封部。
具体实施方式
[0029]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0030]
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
[0031]
本发明实施例中乙烯基乙醚(99%,试剂级)购自acros;
[0032]
本发明实施例中三乙基氧膦(98%,试剂级)购自阿拉丁;
[0033]
本发明实施例中偶氮二异丁腈(98%,试剂级),购自北京伊诺凯科技有限公司;
[0034]
本发明实施例中乙二膦(h2pc2h4ph2化学名1,2-双(膦基)乙烷)为自制,每个实施例和对比例均需要提前制备乙二膦,其制备方法为:
[0035]
合成(c2h5o)2p(o)c2h4(o)p(oc2h5)2(亚乙基二磷酸四乙酯):取314g三乙基氧膦和200给二溴乙烷,置于500ml的烧瓶中,加上高效分馏柱,145-150℃下油浴1.5h,将馏分成分去除,接着将烧瓶中剩余的反应物冷却至室温,然后减压蒸馏,获得155-157℃/1mmhg的(c2h5o)2p(o)c2h4(o)p(oc2h5)
2 62g;
[0036]
获取乙二膦:先向反应瓶内通入干燥的氮气,反应瓶中加入20g四氢铝锂和500ml无水乙醚,接着缓慢滴加50g(c2h5o)2p(o)c2h4(o)p(oc2h5)2和60ml无水乙醚,维持反应体系温度在-2-0℃;滴加完毕后,室温静置过夜,然后边搅拌边缓慢滴加400ml 6n的浓盐酸获得反应物,接着将反应物转移到干净烧瓶中,加入10g无水硫酸钠常温干燥8h,将干燥后的反应物移入500ml烧瓶中,在氮气保护下常压蒸馏获得3g乙二膦,蒸馏温度为114-117℃。
[0037]
实施例1
[0038]
本实施例提供一种反应装置,参照图1,其为反应管,反应管为一体成型的玻璃管。所述反应管包括适于放置反应物的容纳部1和适于熔封的熔封部2,所述熔封部2的长度大于所述容纳部1的长度,熔封部2的长度是容纳部1的长度的4倍。
[0039]
本实施例提供一种双乙氧乙膦烷的制备方法,包括如下步骤:
[0040]
取15ml乙烯基乙醚放入收集瓶中,在收集瓶中充满氮气并密封,制备乙二膦时,直接将蒸馏出来的3g乙二膦接入收集瓶中获得混合溶液;
[0041]
取上述的反应管,将反应管底部浸入冰水浴中,取0.3g偶氮二异丁腈放入反应管中,向反应管中通入氮气至饱和,参照图2,将收集有乙烯基乙醚和乙二膦的收集瓶通过软管与反应管连接,使软管的端部浸入偶氮二异丁腈中,向收集瓶中通入氮气将收集瓶中的混合溶液压入反应管中,然后立即再次向反应管中充入氮气至氮气饱和;
[0042]
将反应管从冰浴条件下取出,将反应管倾斜,使用酒精喷灯在靠近熔封部的上端处加热,加热时不断旋转反应管,反应管软化时,轻拉熔封部上端,使熔封部上端与反应管分离,完成熔封;
[0043]
熔封后将反应管静置冷却,然后将反应管置于75℃油浴反应16h,反应后冷却至室
温,将反应管中的溶液移入50ml蒸馏瓶中,在80℃减压蒸馏1h获得双乙氧乙膦烷。
[0044]
实施例2
[0045]
本实施例提供一种双乙氧乙膦烷的制备方法,制备时所使用的反应装置与实施例1中反应装置的唯一区别在于:熔封部2的长度是容纳部1的长度的3倍,该制备方法包括如下步骤:
[0046]
取15ml乙烯基乙醚放入收集瓶中,在收集瓶中充满氮气并密封,制备乙二膦时,直接将蒸馏出来的3g乙二膦接入收集瓶中获得混合溶液;
[0047]
取反应管,将反应管底部浸入冰水浴中,取0.3g偶氮二异丁腈放入反应管中,向反应管中通入氮气至饱和,参照图2,将收集有乙烯基乙醚和乙二膦的收集瓶通过软管与反应管连接,使软管的端部浸入偶氮二异丁腈中,向收集瓶中通入氮气将收集瓶中的混合溶液压入反应管中;
[0048]
将反应管从冰浴条件下取出,将反应管倾斜,使用酒精喷灯在靠近熔封部的上端处加热,加热时不断旋转反应管,反应管软化时,轻拉熔封部上端,使熔封部上端与反应管分离,完成熔封;
[0049]
熔封后将反应管静置冷却,然后将反应管置于75℃油浴反应16h,反应后冷却至室温,将反应管中的溶液移入50ml蒸馏瓶中,在80℃减压蒸馏1h获得双乙氧乙膦烷。
[0050]
实施例3
[0051]
本实施例提供一种双乙氧乙膦烷的制备方法,制备时所使用的反应装置与实施例1中的反应装置完全相同,该制备方法包括如下步骤:
[0052]
取15ml乙烯基乙醚放入收集瓶中,在收集瓶中充满氮气并密封,制备乙二膦时,直接将蒸馏出来的3g乙二膦接入收集瓶中获得混合溶液;
[0053]
取反应管,将反应管底部浸入冰水浴中,取0.3g偶氮二异丁腈放入反应管中,参照图2,将收集有乙烯基乙醚和乙二膦的收集瓶通过软管与反应管连接,使软管的端部浸入偶氮二异丁腈中,向收集瓶中通入氮气将收集瓶中的混合溶液压入反应管中,然后立即再次向反应管中充入氮气至氮气饱和;
[0054]
将反应管从冰浴条件下取出,将反应管倾斜,使用酒精喷灯在靠近熔封部的上端处加热,加热时不断旋转反应管,反应管软化时,轻拉熔封部上端,使熔封部上端与反应管分离,完成熔封;
[0055]
熔封后将反应管静置冷却,然后将反应管置于75℃油浴反应16h,反应后冷却至室温,将反应管中的溶液移入50ml蒸馏瓶中,在80℃减压蒸馏1h获得双乙氧乙膦烷。
[0056]
实施例4
[0057]
本实施例提供一种双乙氧乙膦烷的制备方法,制备时所使用的反应装置与实施例1中的反应装置完全相同,该制备方法包括如下步骤:
[0058]
取15ml乙烯基乙醚放入收集瓶中,在收集瓶中充满氮气并密封,制备乙二膦时,直接将蒸馏出来的3g乙二膦接入收集瓶中获得混合溶液;
[0059]
取反应管,取0.3g偶氮二异丁腈放入反应管中,向反应管中通入氮气至饱和,参照图2,将收集有乙烯基乙醚和乙二膦的收集瓶通过软管与反应管连接,使软管的端部浸入偶氮二异丁腈中,向收集瓶中通入氮气将收集瓶中的混合溶液压入反应管中,然后立即再次向反应管中充入氮气至氮气饱和;
[0060]
将反应管倾斜,使用酒精喷灯在靠近熔封部的上端处加热,加热时不断旋转反应管,反应管软化时,轻拉熔封部上端,使熔封部上端与反应管分离,完成熔封;
[0061]
熔封后将反应管静置冷却,然后将反应管置于75℃油浴反应16h,反应后冷却至室温,将反应管中的溶液移入50ml蒸馏瓶中,在80℃减压蒸馏1h获得双乙氧乙膦烷。
[0062]
对比例1
[0063]
本对比例提供一种双乙氧乙膦烷的制备方法,包括如下步骤:
[0064]
取15ml乙烯基乙醚放入收集瓶中,在收集瓶中充满氮气并密封,制备乙二膦时,直接将蒸馏出来的3g乙二膦接入收集瓶中获得混合溶液;
[0065]
取带有聚四氟乙烯旋盖的玻璃管,将玻璃管底部浸入冰水浴中,取0.3g偶氮二异丁腈放入玻璃管中,向玻璃管中通入氮气至饱和,使用实施例1中的方法,将收集有乙烯基乙醚和乙二膦的收集瓶通过软管与玻璃管连接,使软管的端部浸入偶氮二异丁腈中,向收集瓶中通入氮气将收集瓶中的混合溶液压入玻璃管中,然后立即再次向玻璃管中充入氮气至氮气饱和,使用聚四氟乙烯材质的旋盖旋紧于玻璃管上进行密闭;
[0066]
然后将玻璃管置于75℃油浴反应16h,反应后冷却至室温,将玻璃管中的溶液移入50ml蒸馏瓶中,在80℃减压蒸馏1h获得双乙氧乙膦烷。
[0067]
试验例
[0068]
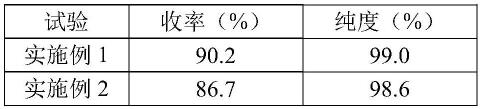
分别按照高效液相色谱法对实施例、对比例制得的双乙氧乙膦烷的纯度进行测试,并按照按照反应摩尔比计算收,收率和纯度见表1。
[0069]
表1.实施例、对比例产物收率和纯度
[0070][0071][0072]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1