一种凝结芽孢杆菌H-1的表达载体及其构建方法与应用
一种凝结芽孢杆菌h-1的表达载体及其构建方法与应用
技术领域
1.本发明涉及基因工程领域,尤其涉及一种基于接合转移方法转化凝结芽孢杆菌h-1的表达载体及其应用。
背景技术:
2.嗜热凝结芽孢杆菌(bacillus coagulans)是芽孢杆菌属的重要成员,是一种革兰氏阳性,兼性厌氧,非致病性,产孢子、产乳酸菌。最佳生长温度为45~60℃,最佳生长ph值6.5~7.0。嗜热凝结芽孢杆菌具有耐热、适合开放式发酵、兼性厌氧、低能耗、高碳代谢速率、高转化率等优势,不仅是重要的益生菌,在工业上也具有重要地位。
3.异源表达系统是改造微生物,拓展微生物菌株应用范围的有力工具,已经在生物催化中被广泛的应用,例如精细化工生产、生物先修复和生物脱硫。合适的异源表达系统不仅可以使微生物的菌株改造或者构建变得容易,可以赋予基因工程菌优良的特性,使得利用生物催化剂进行大规模生产更经济可行。目前,只有很少的基因操作工具可以在凝结芽孢杆菌中工作,而且转化效率极低,很难进行操作。这限制了嗜热凝结芽孢杆菌的优良特性在工业和健康领域的应用。
4.研究基因的作用的常用方法是删除基因,但更多情况下,删除基因组靶标是不可行或不理想的。因为,有的基因是必需基因,细胞生存必不可少;有的细菌基因操作系统仍未完善建立,没有手段进行敲除,比如凝结芽孢杆菌。基因敲低可以实现中等或有条件的抑制。反义rna介导的基因沉默技术目前已广泛用作常规工具。在基因治疗,调控细菌基因表达,质粒复制等领域有广泛应用。可以在不改变基因组的前提下对细菌的关键基因进行调控和研究。真核生物中的小rna(srna)可以特异性敲低各种靶细胞中的基因表达,如sirna和mirna。srna由靶结合序列和hfq伴侣蛋白组成,与之相比反义rna可以单独发挥作用,更为便捷简单。
5.因此,本领域的技术人员致力于开发一种基于接合转移方法转化凝结芽孢杆菌h-1的表达载体及开发一种可以在凝结芽孢杆菌h-1中应用的反义rna工具。
技术实现要素:
6.为实现上述目的,本发明提供了一种基于接合转移方法转化凝结芽孢杆菌h-1的表达载体及其应用,优化了凝结芽孢杆菌h-1的接合转移方法和转化子验证方法,筛选出了四环素和卡那霉素两种可用抗生素及其工作浓度,使用反义rna技术敲弱了甲基转移酶的表达,表现了良好的操作性和较高的转化率,远好于目前的电转化的转化方法。
7.本发明是通过以下技术方案实现的:
8.本发明的第一个方面是提供一种凝结芽孢杆菌h-1的表达载体,包括来源于载体pnw33n的复制子ori、复制子repb,来源于载体pkvm1的dna转移起始位点orit,抗性基因。
9.优选地,所述抗性基因为四环素抗性基因tetr,载体命名为pnwmt,所述载体pnwmt的核苷酸序列如seq id no.1所示。
10.优选地,所述抗性基因为卡那霉素抗性基因kanr,载体命名为pnwmk,所述载体pnwmk的核苷酸序列如seq id no.2所示。
11.进一步,优选地,所述pnwmt载体还包括反义rna表达模块。
12.优选地,所述反义rna表达模块包括启动子p
ldh1
、限制酶识别位点、反义rna序列、终止子tb5和终止子rrnb t1。
13.进一步,优选地,所述反义rna序列以甲基转移酶hsdm846为目标基因,载体命名为pnwmtas,所述载体pnwmtas的核苷酸序列如seq id no.3所示。
14.载体pnwmtas的所述dna元件,其中复制子ori和复制子repb来自载体pnw33n,四环素抗性基因tetr来自载体phy300plk,dna转移起始位点orit来自载体pkvm1。反义rna表达模块来自人工设计合成,其中启动子p
ldh1
来自凝结芽孢杆菌h-1,反义rna序列是目标基因起始密码子前30nt的反义序列,终止子tb5来自枯草芽孢杆菌,终止子rrnb t1来自大肠杆菌
15.本发明的第二个方面是提供所述的表达载体在凝结芽孢杆菌h-1敲弱基因表达中的应用。
16.本发明的第三个方面是提供所述表达载体构建方法,包括以下步骤:
17.s1从载体pnw33n上pcr扩增出复制子ori和复制子repb片段,从载体pkvm1上pcr扩增出dna转移起始位点orit片段,从含有抗性基因的载体上pcr扩增出抗性基因片段;
18.s2将复制子ori和复制子repb片段和dna转移起始位点orit片段进行融合pcr;
19.s3将复制子ori、复制子repb和dna转移起始位点orit片段与抗性基因片段使用无缝克隆技术进行连接,即可得到目的载体;
20.s4将s3获得的目的载体转化到供体菌,再通过结合转移的方法,将目的载体转化到受体菌嗜热凝结芽孢杆菌h-1中。
21.优选地,所述结合转移的方法中,供体菌与受体菌以总od
600
值以10:1的比例混合。
22.优选地,所述供体菌为大肠杆菌s17-1菌株。
23.本发明具有如下有益效果为:
24.1、利用实验室已有的穿梭载体pnw33n,pkvm1,pcas和phy300plk中的各元件,使用接合转移方法转化凝结芽孢杆菌h-1,构建了可以在大肠杆菌和凝结芽孢杆菌h-1中自主复制的载体pnwmt和pnwmk。并使用反义rna表达模块研究了载体的可用性和在反义rna技术的应用。
25.2、以pnwm质粒为基础,探索出高效准确的接合转移转化凝结芽孢杆菌h-1的方法和转化子的验证方法。在探索可用抗生素抗性基因的过程中,得到了携带四环素抗性基因的pnwmt质粒和携带卡那霉素抗性基因的的pnwmk质粒,摸索出了可以在凝结芽孢杆菌h-1中正常工作的两种抗生素及其工作浓度。基本完成了凝结芽孢杆菌h-1的基础基因操作工具。并以反义rna技术敲弱凝结芽孢杆菌h-1dna甲基转移酶hsdm846表达对其生长的影响为实例,考察了所构建载体在反义rna技术应用中的优势。首先用本发明的载体pnwmt构建了对照菌株h1c,用载体pnwmtas构建了敲弱dna甲基转移酶hsdm846表达的工程菌h1as846,使用这两株菌与野生h-1在活化两代的情况下测量其在50℃条件下的生长情况,发现h1as846的生长被严重抑制。这表明本发明所构建的载体的实用性以及反义rna表达模块在凝结芽孢杆菌h-1中的实用性,在对关键基因的研究上有更大的应用价值。
附图说明
26.图1是载体pnwmt的构建示意图;
27.图2是pcr验证载体pnwmt的电泳图(m1:分子标记物;o1:以载体pnwmt为模板,orirep.f(kpnⅰ)和orirep.r为引物进行pcr扩增的结果;j1:以载体pnwmt为模板,mob.f和mob.r为引物进行pcr扩增的结果;t1:以载体pnwmt为模板,tetr.f和tetr.r(kpnⅰ)为引物进行pcr扩增的结果);
28.图3是载体pnwmk的构建示意图;
29.图4是pcr验证载体pnwmk的电泳图(m2:分子标记物;c2:对照;a2:以载体pnwmk为模板,kanr.f和kanr.r为引物进行pcr扩增的结果)
30.图5是载体pnwmtas的构建示意图;
31.图6是pcr验证载体pnwmtas的电泳图(m1:分子标记物;c2:对照;a2:以载体pnwmtas为模板,asrna.f和asrna.r为引物进行pcr扩增的结果);
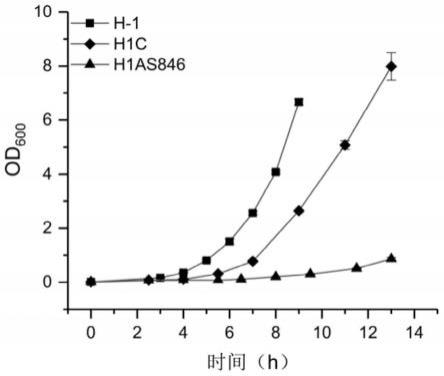
32.图7是反义rna敲弱dna甲基转移酶hsdm846对生长的影响曲线图。
具体实施方式
33.以下参考说明书附图介绍本发明的多个优选实施例,使其技术内容更加清楚和便于理解。本发明可以通过许多不同形式的实施例来得以体现,本发明的保护范围并非仅限于文中提到的实施例。
34.以下实施例中未注明具体方法的实验方法,按照常规方法,具体参见《分子克隆实验手册》(new york:cold spring harbor laboratory press,1989)中所述的条件,或者按照试剂和试剂盒制造厂商所建议的条件。
35.本发明所有用到的dna元件和微生物菌株均来自已经公开发表文献资料,其序列信息和dna本身均可以被公众获得,详见表1和表2;其他各种材料均系本发明构建,可以用本发明所述的方法进行再构造;
36.表1本实施例涉及的菌株列表
[0037][0038]
表2本实施例涉及的载体列表
[0039][0040]
表3本实施例涉及的引物列表
[0041]
[0042][0043]
注:小写字母表示前后片段同源序列
[0044]
实施例1通过接合转移转化凝结芽孢杆菌h-1的表达载体pnwmt的构建
[0045]
质粒pnw33n是一种可以在大肠杆菌和枯草芽孢杆菌中表达的穿梭质粒,也被用于电转化凝结芽孢杆菌,但是目前电转化的效率极低,很难通过电转化稳定得到改造菌株。本实施例通过提取载体pnw33n的复制子ori和复制子repb,将其与载体pkvm1的dna转移起始位点orit,载体phy300plk的四环素抗性基因tetr组合在一起,得到通过接合转移转化凝结芽孢杆菌h-1的表达载体pnwmt。
[0046]
为此,首先以orirep.f(kpnⅰ)和orirep.r为引物从载体pnw33n上pcr扩增出复制子ori和复制子repb片段,以mob.f和mob.r为引物从载体载体pkvm1上pcr扩增出dna转移起始位点orit片段,以tetr.f和tetr.r为引物从载体phy300plk上pcr扩增出四环素抗性基因tetr片段。然后,用引物orirep.f(kpnⅰ)和mob.r为引物将复制子ori和复制子repb片段和dna转移起始位点orit片段进行融合pcr。最后将复制子ori、复制子repb和dna转移起始位点orit片段与四环素抗性基因tetr片段使用无缝克隆技术进行连接,即可得到目的载体pnwmt(图1)
[0047]
在载体构建的过程中以四环素抗性作为四环素抗性基因是否插入的活性指标,构建好的载体使用pcr扩增的方法提供生物分子学证据证明是否正确。图2是用pcr方法对载
体pnwmt进行验证的结果,由图可知用引物orirep.f(kpnⅰ)和orirep.r可以扩增出2000~2500bp的条带o1,这和克隆的复制子ori和复制子repb片段大小相符。用引物mob.f和mob.r可以扩增出1600~2000bp的条带j1,这和克隆的dna转移起始位点orit片段大小相符。用引物tetr.f和tetr.r可以扩增2000bp大小的条带t1,这与四环素抗性基因tetr片段大小相符。说明载体pnwmt的构建是成功的。
[0048]
进一步验证采用测序的方法,使用dna测序的方法对对载体pnwmt最终进行验证,验证结果表明所测得的构建载体的序列和预计的完全一致。
[0049]
实施例2通过接合转移转化凝结芽孢杆菌h-1的表达载体pnwmk的构建
[0050]
在异源化改造微生物系统的过程中,抗生素抗性基因能起到筛选目标菌株和外缘压力维持质粒等重要作用。对于一个未曾进行基因操作的菌种,多开发可用的抗性基因有重要作用,可以为后续基因操作如crispr双质粒系统等的开发铺平道路。本实施例以pnwmt为基本骨架,开发出了第二种可以在嗜热凝结芽孢杆菌h-1中使用的抗性基因卡那霉素抗性基因。本实施例通过提取载体pnwmt的复制子ori和复制子repb,dna转移起始位点orit与载体pcas的卡那霉素抗性基因kanr组合在一起,得到通过接合转移转化凝结芽孢杆菌的表达载体pnwmk(图3)。为此,首先以orirepmob.f和orirepmob.r为引物从载体pnwmt上pcr扩增出复制子ori、复制子repb和dna转移起始位点orit片段,以kanr.f和kanr.r为引物从载体pcas上pcr扩增出卡那霉素抗性基因kanr片段。将复制子ori、复制子repb和dna转移起始位点orit片段与卡那霉素抗性基因kanr片段使用无缝克隆技术进行连接,即可得到目的载体pnwmtk(图3)
[0051]
在载体构建的过程中以卡那霉素抗性作为卡那霉素抗性基因是否插入的活性指标,构建好的载体使用pcr扩增的方法提供生物分子学证据证明是否正确。图4是用pcr方法对载体pnwmk进行验证的结果,由图可知用pnwmt作为模板(对照)时不能用引物kanr.f和kanr.r扩增出条带。用pnwmk作为模板用引物kanr.f和kanr.r可以扩增1000~1200bp大小的条带t1,这与卡那霉素抗性基因kanr片段大小相符。说明载体pnwmk的构建是成功的。
[0052]
进一步验证采用测序的方法,使用dna测序的方法对对载体pnwmk最终进行验证,验证结果表明所测得的构建载体的序列和预计的完全一致。
[0053]
实施例3培养方法
[0054]
嗜热凝结芽孢杆菌是一种高效的生产乳酸的菌株,具有耐热、适合开放式发酵、兼性厌氧、低能耗、高碳代谢速率、高转化率等优势。嗜热凝结芽孢杆菌h-1具有更强的产酸能力,其培养基中需含有适量的葡萄糖才能正常生长,而且在普通的lb等无缓冲剂培养基里不能正常生长。经过探索,在gys培养基中凝结芽孢杆菌h-1可以正常生长。gys培养基的配方是每升蒸馏水中加入100g葡萄糖,50g轻质碳酸钙,10g酵母粉和5g蛋白胨。适宜的培养温度是50℃。在一些操作中需要降低轻质碳酸钙的干扰,比如接合转移,三代测序样品制备等。gys培养基中轻质碳酸钙的含量可以适当降低。经测试gys5培养基可以让凝结芽孢杆菌h-1正常生长到od
600
值为2,可以满足接合转移,三代测序样品制备等操作的需求。gys5培养基的配方是每升蒸馏水中加入100g葡萄糖,5g轻质碳酸钙,10g酵母粉和5g蛋白胨。
[0055]
实施例4优化的接合转移方法
[0056]
常见的接合转移方法并不适合嗜热凝结芽孢杆菌,本实施例根据凝结芽孢杆菌的生理性质将接合转移方法进行优化,得到了较高转化率。本实施例将表达载体pnwmt化学转
化到大肠杆菌s17-1菌株中做为质粒供体菌,将从单菌落接种到液体培养基并活化两代的凝结芽孢杆菌作为受体菌。将大肠杆菌s17-1以od
600
为0.01的初始浓度接种于浓度为25ng/ml四环素的lb培养基培养至对数期初期,即od
600
在0.5~1。凝结芽孢杆菌h-1接种于gys5培养基(初始od
600
为0.06),培养至od
600
在0.5~1(约4.5h)。通过离心(2min,5000
×
g,37℃)收获细胞。大肠杆菌用37℃预热的lb培养基洗涤两遍,凝结芽孢杆菌h-1用37℃预热的lb培养基洗涤一遍。将供体菌与受体菌以总od
600
值以10:1的比例混合,离心(2min,5000
×
g,37℃)沉淀。吸去多余的培养基,使用20μl lb培养基轻柔的重悬菌体,滴在37℃预热的无抗生素的固体lb平板上,吹干后在37℃培养箱中正面放置孵育4h。孵育结束后将菌体从平板上刮下,在1ml无碳酸钙的gys培养基中重悬,涂布在含2.5μg/ml四环素的固体gys培养基平板上。50℃条件下避光孵育,两天后即可得到单菌落。
[0057]
实施例5优化的转化子的验证方法
[0058]
嗜热凝结芽孢杆菌h-1在接合转移的转化子的验证方法和普通的大肠杆菌或芽孢杆菌不同。因为其只能在含有轻质碳酸钙的培养基中培养且生长速度较慢,导致菌液pcr验证操作麻烦且效率低下。嗜热凝结芽孢杆菌h-1的初代转化子中还有很强的丢失质粒的现象,会有不少的转化子验证正确但接种培养后质粒丢失。
[0059]
本实施例研究出了一种方便准确的转化子验证方法。挑取需要验证的转化子单菌落,在20μl的lb培养基中混匀。吸取1μl用于pcr验证,如果存在相应的条带,则把该转化子对应的剩余19μl菌液在含抗生素的gys固体培养基上划线,50℃条件下避光孵育两天。选择生长良好的单菌落进行pcr验证,如果条带正确测序结果正确,那么这就是可以稳定保存工程质粒的目标菌株。
[0060]
实施例6嗜热凝结芽孢杆菌h-1适合的四环素抗生素工作浓度
[0061]
嗜热凝结芽孢杆菌h-1进行接合转移后,我们发现其可以正常生长的抗生素浓度与大肠杆菌的并不一致。嗜热凝结芽孢杆菌h-1适合的抗生素工作浓度远小于大肠杆菌所需。
[0062]
本实施例验证了嗜热凝结芽孢杆菌h-1的适宜四环素抗生素工作浓度。制作一批四环素抗性的gys固体培养基,其四环素的浓度设置梯度为50,25,12.5,5,2.5和1.25μg/ml。按照实施例4中的方法将质粒pnwmt转入凝结芽孢杆菌h-1中,涂布在梯度浓度四环素的gys固体培养基中,50℃条件下避光孵育,观察转化子生长情况。继续按照实施例5中的方法筛选出pcr阳性的初代转化子在其对应浓度的四环素gys固体培养基上划线接种,50℃条件下避光孵育,观察生长情况并挑取单菌落进行pcr验证。
[0063]
具体结果在表3中,可见5或2.5μg/ml是嗜热凝结芽孢杆菌h-1对应的四环素抗生素正常工作浓度,在该浓度下质粒可以在抗生素压力下稳定存在,而且对细菌的生长影响较小。
[0064]
表4嗜热凝结芽孢杆菌h-1四环素抗生素工作浓度
[0065][0066]
实施例7
[0067]
适合的卡那霉素抗生素浓度
[0068]
嗜热凝结芽孢杆菌h-1进行接合转移后,我们发现其可以正常生长的抗生素浓度与大肠杆菌的并不一致。嗜热凝结芽孢杆菌h-1适合的抗生素工作浓度远小于大肠杆菌所需。本实施例验证了嗜热凝结芽孢杆菌h-1的适宜卡那霉素抗生素工作浓度。制作一批卡那霉素抗性的gys固体培养基,其四环素的浓度设置梯度为200,100,50,25,12.5和5μg/ml。按照实施例4中的方法将质粒pnwmk转入凝结芽孢杆菌h-1中,涂布在梯度浓度卡那霉素的gys固体培养基中,50℃条件下孵育,观察转化子生长情况。继续按照实施例5中的方法筛选出pcr阳性的初代转化子在其对应浓度的卡那霉素gys固体培养基上划线接种,50℃条件下孵育,观察生长情况并挑取单菌落进行pcr验证。具体结果在表4中,可见25μg/ml是嗜热凝结芽孢杆菌h-1对应的卡那霉素抗生素正常工作浓度,在该浓度下质粒可以在抗生素压力下稳定存在,卡那霉素对细菌生长影响较小,即使在50μg/ml浓度下,初代转化子也仅需两天就能孵育出来。
[0069]
表5嗜热凝结芽孢杆菌h-1卡那霉素抗生素工作浓度
[0070][0071]
实施例8通过接合转移转化凝结芽孢杆菌反义rna表达载体pnwmtas的构建
[0072]
反义rna介导的基因沉默技术目前已广泛用作常规工具。在基因治疗,调控细菌基因表达,质粒复制等领域有广泛应用。可以在不改变基因组的前提下对细菌的关键基因进行调控和研究。因此有必要开发一种可以在凝结芽孢杆菌h-1中应用的反义rna工具。甲基转移酶是dna甲基化的关键酶,dna甲基化是表观遗传的主要机制。凝结芽孢杆菌是dna甲基化程度较高的菌种。凝结芽孢杆菌h-1表达量最高的dna甲基转移酶hsdm846,直接敲除dna
甲基化酶的操作难度很大。本实施例以甲基转移酶hsdm846为反义rna目标基因,反义rna序列为5
‘‑
tcttgaagtttggttcctcctttttcttac-3’。p
ldh1
启动子是凝结芽孢杆菌h-1中极高表达的l乳酸脱氢酶基因的启动子,是反义rna表达模块的优良启动子。本实施例用p
ldh1
启动子来表达asrna,插入载体pnwmt构建了可以通过接合转移转化凝结芽孢杆菌h-1的反义rna表达载体pnwmtas(图5)。为此,首先以orirepmob.f和orirepmob.r从载体pnwmt上pcr扩增出复制子ori、复制子repb和dna转移起始位点orit片段,以asrna.f和asrna.r为引物扩增出反义rna表达模块,以tetras.f和tetras.r为引物从载体phy300plk上pcr扩增出四环素抗性基因tetr片段。然后,以asrna.f和tetras.r为引物将反义rna表达模块和四环素抗性基因tetr片段进行融合pcr。最后将复制子ori、复制子repb和dna转移起始位点orit线性片段与反义rna表达模块和四环素抗性基因tetr线性片段使用无缝克隆方法进行连接,即可得到目的载体pnwmtas(图5)。
[0073]
构建好的载体使用pcr扩增的方法提供生物分子学证据证明是否正确。图6是用pcr方法对载体pnwmt进行验证的结果,由图可知用pnwmt作为模板(对照)时不能用引物asrna.f和asrna.r扩增出条带,用pnwmtas作为模板时可以扩增出800bp大小的条带,这与反义rna表达模块大小相符。这一结果说明已经成功构建了载体pnwmtas。
[0074]
进一步验证采用测序的方法,使用dna测序的方法对对载体pnwmt最终进行验证,验证结果表明所测得的构建载体的序列和预计的完全一致。
[0075]
实施例9接合转移方法
[0076]
本实施例将表达载体pnwmt和pnwmtas通过接合转移方法转化凝结芽孢杆菌h-1,具体的接合转移方法和实施例4相同。由于pnwmtas其中的反义rna元件对凝结芽孢杆菌h-1的生理状态有影响,需要扩大接合转移的细菌量才能得到较多的转化子,合适的凝结芽孢杆菌受体菌的量为10~15ml。使用载体pnwmt接合转移转化凝结芽孢杆菌得到转化子h1c,使用载体pnwmtas接合转移转化凝结芽孢杆菌得到转化子h1as846。
[0077]
实施例10反义rna敲弱甲基转移酶hsdm846对凝结芽孢杆菌的生长情况的影响
[0078]
dna甲基化是表观遗传的主要机制,能引起染色质结构、dna构象、dna稳定性及dna与蛋白质相互作用方式的改变,从而控制基因表达、dna错配修复和转座。hsdm846是凝结芽孢杆菌h-1表达量最高的dna甲基转移酶,它的敲弱可能会对细菌的生长产生较大影响。本实施例以h1as846菌株为实验菌株,h1c为实验对照菌株,原始的h-1菌株为空白对照菌株。挑取以上菌株的单菌落,接种于30ml gys培养基中过夜培养。以初始od
600
为0.015的条件接种至新的gys培养基中,作为种子菌株继续培养,在od
600
为0.5~1时接种至新的gys培养基中(初始od
600
为0.015)。每1.5~2h取样测量od
600
值并绘制生长曲线,以上过程的培养温度均为50℃,h1as846菌株和h1c菌株的培养过程中的培养基均含有浓度为2.5μg/ml的四环素。结果显示如图5所示。菌株h1c相比于h-1菌株的生长显著放缓。菌株h1as846的生长速度变得非常慢,其需要约14h才能生长到od
600
等于1。这表明反义rna技术在凝结芽孢杆菌中起到了敲弱甲基转移酶hsdm846表达的效果,载体pnwmtas可以有效敲弱凝结芽孢杆菌中的基因表达。甲基化酶hsdm846是影响凝结芽孢杆菌h-1生长的关键dna甲基化酶。因此,除上述证明的优势外,本实施例构建的通过接合转移转化凝结芽孢杆菌h-1的反义rna表达载体还具有调控基因表达的优势,它可以使得对关键基因的调控及研究成为可能。
[0079]
以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术无需创
造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1