一种改进的酮咯酸氨丁三醇中间体的制备方法与流程
1.本发明属于药物化学领域,具体涉及一种改进的酮咯酸的制备方法,所述酮咯酸是制备酮咯酸氨丁三醇的中间体。
背景技术:
2.酮咯酸氨丁三醇是一种新型的可供注射的非甾体类强力止痛及中度抗炎解热药,其作用机理是通过抑制机体外周和中枢前列腺素(pg)的产生,从而降低外周和中枢中的pg,其主要成分酮咯酸的化学结构如下:
[0003][0004]
目前,合成酮咯酸氨丁三醇的主要方法有两种,其一主要是2-苯甲酰吡咯为起始物料经过氧化缩合、合环、脱酸,最后成盐得到酮咯酸氨丁三醇(cn114031621;中国药物化学杂志,1995,5(3):223-225),其反应路线如下:
[0005][0006]
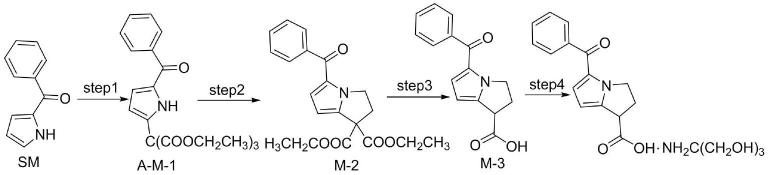
其二主要也是2-苯甲酰吡咯为起始物料,经过取代、合环、脱羧,最后得到酮咯酸氨丁三醇(中国医药工业杂志,2019,50(8):868-870;中国药物化学杂志,2002,12(4):228-229),其反应路线如下:
[0007][0008]
在合成酮咯酸氨丁三醇的两条路线中,均存在m-2水解脱羧合成酮咯酸的过程,说明酮咯酸的合成尤为重要。然而,由于酮咯酸性质不稳定,易被氧化产生有色杂质,严重影响后续制剂的质量和安全。在中国药典2020年版二部中对酮咯酸氨丁三醇的原料药的溶液颜色进行了规定,限制其溶液的颜色不得深于黄色或黄绿色3号标准比色液。同时,酮咯酸氨丁三醇注射液的颜色也进行了限制。表明酮咯酸丁三醇的颜色杂质对于制剂的安全具有重要的影响。
[0009]
然而,现有文献公布的合成步骤中大多采用加入还原剂进行抑制氧化颜色杂质的产生,专利cn101575340b在酮咯酸成盐的步骤加入含硫的无机盐还原剂,而专利cn114349757a则在水解脱羧步骤加入含硼的还原剂。前者存在增加无机盐带入酮咯酸氨丁
三醇成品的风险,而后者采用含硼的还原剂直接加入碱液中,不仅体系会产生大量的氢气,给大生产带来较大的安全隐患,而且生成的亚硼酸钠易溶于水,导致后续得到的中间体m-3中仍然存在引入无机盐的风险。同时,两者后处理方式仍然都采用浓缩后调酸碱而后再次纯化的方式,长时间的减压浓缩不仅会增加不稳定酮咯酸产生其它杂质的可能,而且繁琐的操作也增加了大生产的周期和能源的成本。
[0010]
针对现有技术存在问题,本发明人经过大量的试验,意外地发现了影响酮咯酸氨丁三醇颜色杂质产生的根源在于m-3的生产过程产生的颜色杂质,而控制m-3生产过程颜色杂质的产生主要在于水解脱羧的全过程。因此,在专利cn114031621的方法的基础上对于水解脱羧的反应和后处理过程进行了研究,发现在反应中严格控制反应体系溶液中的含氧量,同时,在后处理过程中,特别是调酸碱过程中严格控制氧气对于m-3生成的影响,上述措施可较好地控制m-3合成过程中颜色杂质的产生。同时,水解脱羧反应采用两相反应,极大地利用了两相反应的优势,避免了现有技术不得不长时间浓缩的后处理存在的弊端,从而减少了因长时间加热溶液导致m-3相关杂质产生的概率。另外,本发明人在试验中也意外地发现醚类化合物对于颜色的去除也具有明显的作用,过滤得到的m-3湿品不经过任何处理,直接采用醚类化合物打浆处理就可以达到现有技术采用醇类和水纯化的效果。同时,也避免了现有技术中因加入含硫或硼还原性化合物带来的无机盐难以去除的技术问题及后续还需要再用醇类和水进行纯化的繁琐步骤。通过对反应过程控氧和采用醚类化合物进行后处理获得的m-3经过简单的脱色、成盐转化成酮咯酸氨丁三醇(制备方法参考cn101143865,全文引入,不再赘述。)。按照中国药典溶液颜色检查法进行检查,得到的酮咯酸氨丁三醇溶液颜色均浅于黄绿色1号标准比色液,炽灼残渣均在0.1%以下。其hplc检测纯度大于或等于99.7%,单个最大杂质均在0.10%以下,符合药用的要求,从而完成本发明。
技术实现要素:
[0011]
本发明的目的在于提供一种改进的酮咯酸氨丁三醇中间体(即酮咯酸)的制备方法,该方法可有效减少有色杂质的产生,从而终品酮咯酸氨丁三醇的颜色杂质控制在中国药典规定的范围。
[0012]
为实现本发明的目的,提供如下实施方案。
[0013]
在一实施方案中,本发明的一种改进的酮咯酸氨丁三醇中间体的制备方法,包括将式m-2化合物溶于有机溶剂,在碱溶液存在下进行水解脱羧反应,分离获得的产物加入到醚类化合物中打浆处理,过滤,干燥即得到式m-3的酮咯酸,
[0014][0015]
其中,所述碱液预先脱氧,在所述反应和打浆处理过程中均采用惰性气体保护。
[0016]
上述本发明的制备方法,具体包含以下步骤:
[0017]
1)碱溶液配制:将固体无机碱溶解于预先脱氧的水中;
[0018]
2)将式m-2化合物溶于有机溶剂中,加入上步的碱溶液、相转移催化剂,通入惰性气体保护;
[0019]
3)加热升温至35~45℃反应,反应完后分液,取水相用乙酸乙酯洗涤两次;
[0020]
4)在惰性气体保护条件下,将水相降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤;
[0021]
5)将滤饼加入到醚类化合物中打浆处理,过滤,固体真空干燥即得酮咯酸。
[0022]
上述本发明的制备方法,所述碱溶液为氢氧化钠溶液或氢氧化钾溶液,所述有机溶剂为甲苯、1,2-二氯乙烷或它们的混合物,所述相转移催化剂为四丁基溴化铵或四丁基氯化铵,所述惰性气体为氮气或者氩气,所述醚类化合物为异丙醚、甲基叔丁基醚或它们的混合物。
[0023]
上述本发明的制备方法,所述碱液预先脱氧,包括将固体无机碱溶解于预先脱氧的水中,或将固体无机碱溶解于水中再脱氧。
[0024]
上述本发明的制备方法,进一步包括将所制得的酮咯酸转化成酮咯酸氨丁三醇盐。按照中国药典溶液颜色检查法进行检查,得到的酮咯酸氨丁三醇溶液颜色均浅于黄绿色1号标准比色液,炽灼残渣均在0.1%以下。其hplc检测纯度大于或等于99.7%,单个最大杂质均在0.10%以下,符合药用的要求。
[0025]
在一具体实施方案中,本发明的一种改进的酮咯酸氨丁三醇中间体的制备方法,包括将m-2化合物溶于有机溶剂甲苯或1,2-二氯乙烷或者两者的混合溶剂中得到的溶液,预先脱氧的碱溶液、相转移催化剂加入到反应容器中,在惰性气体条件下进行升温至35~45℃反应。反应完毕后,分液,水相用乙酸乙酯洗涤两次,在惰性气体保护下,用盐酸调节ph值,过滤得到的m-3湿品,加入到醚类化合物中打浆处理,过滤、滤饼干燥,最后得到m-3的酮咯酸。其中m-2的制备参考cn114031621,全文引入,不再赘述,所述预先脱氧的碱溶液是将配制碱溶液的水预先脱氧。
[0026]
在一具体实施方案中,本发明的一种改进的酮咯酸氨丁三醇中间体的制备方法,具体包含以下步骤:
[0027]
1)将m-2的甲苯和1,2-二氯乙烷的混合溶液、碱液、相转移催化剂加入到反应容器中,惰性气体保护;
[0028]
2)升温至35~45℃反应完全,分液,水相用乙酸乙酯洗涤两次;
[0029]
3)惰性气体保护条件下,降温,用盐酸调ph值1~2;
[0030]
4)过滤,滤饼用水洗涤,得到的m-3湿品直接加入醚类化合物打浆处理,过滤,真空干燥即得m-3粗品。
[0031]
优选的,上述本发明的制备方法,所述液碱为氢氧化钠溶液或氢氧化钾溶液,其用水为预先脱氧纯化水。
[0032]
优选的,上述本发明的制备方法,所述相转移催化剂为四丁基溴化铵或四丁基氯化铵。
[0033]
优选的,上述本发明的制备方法,所述惰性气体为氮气或氩气,更优选地,所述惰性气体为氮气。
[0034]
优选的,上述本发明的制备方法,所述醚类化合物为异丙醚或甲基叔丁基醚或者两者的混合物。更优选地,所述醚类化合物为异丙醚。
[0035]
优选的,上述本发明的制备方法,步骤1)所述相转移催化剂为四丁基溴化铵,四丁
基溴化铵与m-2摩尔比约为0.05:1。所述碱液的浓度为20%~30%。
[0036]
优选的,在一具体实施方案中,本发明的一种改进的酮咯酸氨丁三醇中间体的制备方法,包含以下步骤:
[0037]
1)将含有m-2、甲苯和1,2-二氯乙烷的混合溶液、脱氧的20%氢氧化钠溶液、四丁基溴化铵加入到反应容器中,加氮气保护;
[0038]
2)升温至35~45℃反应完全,分液,水相用乙酸乙酯洗涤两次;
[0039]
3)在氮气保护条件下,水相降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,得到的m-3湿品;
[0040]
4)将m-3湿品直接加入异丙醚中打浆处理,过滤,滤饼真空干燥即得m-3的酮咯酸。
[0041]
术语,所谓“浓度”是指每l水所含碱的重量kg,即kg/l,或g/ml;比如20%氢氧化钠是100ml水中溶解20g氢氧化钠。
[0042]
依据本发明改进的制备方法得到酮咯酸氨丁三醇中间体m-3,避免了现有技术因额外加入含硫或含硼还原剂增加成品中带入无机盐的风险,解决了酮咯酸氨丁三醇中存在的颜色杂质难以解决的难题。本发明的方法操作简单、成本低、纯度高。如下述实施例制得的m-3经过简单的脱色、成盐,即可获得到酮咯酸氨丁三醇成品。按照中国药典溶液颜色检查法进行检查,其成品溶液的颜色均浅于黄绿色1号标准比色液,炽灼残渣均在0.1%以下。其hplc检测纯度大于或等于99.7%,单个最大杂质均在0.10%以下,符合药用的要求。
附图说明
[0043]
图1为对照例1获得的酮咯酸氨丁三醇的hplc图谱;
[0044]
图2为对照例2获得的酮咯酸氨丁三醇的hplc图谱;
[0045]
图3为对照例3获得的酮咯酸氨丁三醇的hplc图谱;
[0046]
图4为实施例1获得的酮咯酸氨丁三醇的hplc图谱。
具体实施方式
[0047]
以下实施例仅是典型的,用于对本发明进行进一步说明和理解,但不以任何形式限制本发明的范围。
[0048]
液相色谱仪:岛津lc-2030c(3d plus);
[0049]
检测器:uv+dad;
[0050]
色谱柱:岛津shimnex cs(4.6*250mm,5μm);
[0051]
流动相:0.5mol/l磷酸二氢铵溶液(3.0)作为流动相a,四氢呋喃作为流动相b,梯度洗脱(0min,85vol.%a-15vol.%b;5min,85vol.%a-15vol.%b;15min,70vol.%a-30vol.%b;30min,60vol.%a-40vol.%b;45min,85vol.%a-15vol.%b;60min,85vol.%a-15vol.%b;);
[0052]
柱温:40℃;
[0053]
流速:1.0ml/min;
[0054]
波长:313nm;
[0055]
进样量:20μl。
[0056]
对照例1酮咯酸氨丁三醇的制备(m-2制备参考中国医药工业杂志,2019,50(8):
868-870,全文引入,不再赘述。)
[0057]
取m-2(50g,0.141mol,1.0equiv.),加入甲醇500ml,30%氢氧化钠溶液(225g,1.69mol,12equiv.),加热至50~60℃搅拌反应完全,加入硼氢化钠(53.2mg,1.407mmol,0.01equiv.),减压蒸出甲醇,降温至0~10℃,滴加浓盐酸调节ph值至2,抽滤,滤饼用1mol/l的稀盐酸250ml打浆,过滤,然后用纯化水100ml淋洗滤饼,真空40
±
5℃干燥后得到30.4g m-3。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色4号和5号标准比色液之间。
[0058]
将m-3(10g,0.039mol,1.0equiv.)加入到95%乙醇30ml中,搅拌加热升温至50-60℃,待固体溶解后,加入活性炭1g,搅拌脱色45分钟,过滤。滤液中加入氨丁三醇(5.7g,0.047mol,1.2equiv.),加热至50~60℃,固体全部溶解,停止加热,将溶液冷却至40℃以下,加入少量晶种,有固体析出,将温度降温至20℃以下,搅拌析晶8小时以上,过滤,滤饼用无水乙醇5ml
×
2淋洗,50
±
5℃真空干燥,得到10.8g酮咯酸氨丁三醇。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色2号和3号标准比色液之间,炽灼残渣0.18%,其纯度99.76%,单个最大杂质0.07%。结果见表1。
[0059]
表1所得酮咯酸氨丁三醇的hplc检测结果
[0060]
峰号保留时间面积面积%高度ntp(usp)分离度(usp)拖尾因子rrt18.00013200.006785725
‑‑
1.30.29214.86028070.0123456588121.131.10.54316.951158440.0661066502617.902.90.61418.41742110.018170158563.30
‑‑
0.66520.78381240.034632586375.101.00.75622.89822600.009133511595.65
‑‑
0.83725.54846070.019226369185.661.00.92827.7392379515499.7601186735432284.111.01.00934.71213660.00610214378815.500.91.251038.10158190.0242761462428.860.71.371139.15858130.0243771418132.591.01.411242.17049860.0212751151046.601.01.52总计 23852310100.0001190416
ꢀꢀꢀꢀ
[0061]
对照例2酮咯酸氨丁三醇的制备(m-2制备参考中国医药工业杂志,2019,50(8):868-870,全文引入,不再赘述。)
[0062]
取m-2(50g,0.141mol,1.0equiv.),加入95%乙醇35ml,10%氢氧化钠溶液18ml,加热至70℃搅拌反应完全,减压蒸出甲醇,-5℃水层用浓盐酸调节ph值至2,抽滤,滤饼用甲醇∶水=3∶1(体积比)精制,得到31.6g m-3。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色3号和4号标准比色液之间。
[0063]
取m-3(10g,0.039mol,1.0equiv.)、氨丁三醇(4.7g,0.039mol,1.0equiv.)、亚硫酸钠0.017g加入59ml甲醇中,加热回流至固体完全溶解。加入0.69g活性炭,加热脱色15分钟,过滤,自然降温析晶,过滤,真空干燥,得到8.7g酮咯酸氨丁三醇。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色1号和2号标准比色液之间,炽灼残渣0.15%,其纯度99.75%,单个最大杂质0.06%。结果见表2。
[0064]
表2所得酮咯酸氨丁三醇的hplc检测结果
[0065]
峰号保留时间面积面积%高度ntp(usp)分离度(usp)拖尾因子rrt18.00110280.004676457
‑‑
1.30.29214.86115740.0062027229922.151.10.54316.961147550.059925470757.873.10.61418.16428850.011120158872.71
‑‑
0.65519.10110130.00461329281.881.20.69620.79077400.031619594734.431.00.75722.26215030.00696445373.86
‑‑
0.80822.89983380.033527500201.53
‑‑
0.83925.55651530.020255367915.641.00.921027.7472510685599.7481257085434764.111.01.001134.71320660.00815614177515.46
‑‑
1.251238.11870800.0282841583049.060.71.371339.17664780.0264251436332.661.01.411442.20737430.0152111209506.741.11.52总计 25170222100.0001261032
ꢀꢀꢀꢀ
[0066]
对照例3酮咯酸氨丁三醇的制备(参考cn114031621,全文引入,不再赘述。)
[0067]
将m-2的甲苯、1,2-二氯乙烷混合溶液(以sm计,0.234mol,1.0equiv.),四丁基溴化铵3.76g(0.012mol,0.05equiv.),氢氧化钠水溶液约233ml(将46.7g氢氧化钠(1.167mol,0.05equiv.)加入到233ml纯化水中进行溶解配置)加入到反应器中。加热至35~45℃反应完全,分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,真空干燥即可得到m-3 54.8g。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色4号和5号标准比色液之间。
[0068]
将m-3按照cn101143865进行制备,得到的酮咯酸氨丁三醇按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色2号和3号标准比色液之间,炽灼残渣0.06%,其纯度99.80%,单个最大杂质0.06%。结果见表3。
[0069]
表3所得酮咯酸氨丁三醇的hplc检测结果
[0070]
峰号保留时间面积面积%高度ntp(usp)分离度(usp)拖尾因子rrt18.05410760.004818057
‑‑
1.40.29214.84215420.0062017362423.501.10.54316.845144190.0581246593478.091.70.61418.41623630.009104637375.530.70.66520.66557150.023454585707.101.00.75622.19219510.00892415853.93
‑‑
0.80722.85365940.026413496271.560.80.83825.54635680.014165303475.401.00.92927.7012494312499.8031243614431883.851.01.001034.75211500.0058614827415.771.01.251138.10736000.0141251294118.550.81.38
1239.15935220.0142321419912.5
‑‑
1.411342.21338580.0152171188616.751.11.52总计 24992463100.0001247030
ꢀꢀꢀꢀ
[0071]
实施例1(m-2制备参考cn1l4031621,全文引入,不再赘述。)
[0072]
合成路线:
[0073][0074]
将m-2的甲苯、1,2-二氯乙烷混合溶液(以sm计,0.234mol,1.0equiv.),四丁基溴化铵3.76g(0.012mol,0.05equiv.),氢氧化钠水溶液约233ml(将46.7g氢氧化钠(1.167mol,0.5equiv.)加入到233ml纯化水中进行溶解配置,纯化水预先进行脱氧)加入到反应器中,氮气保护。加热至35~45℃反应完全,分液,水相用乙酸乙酯洗涤两次。氮气保护下,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,得到的m-3湿品直接加入异丙醚化合物打浆处理,过滤,真空干燥即可得到m-3 55.7g。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色2号和3号标准比色液之间。
[0075]
将m-3按照cn101143865进行制备,得到的酮咯酸氨丁三醇按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色0.5号和1号标准比色液之间,炽灼残渣0.08%,其纯度99.73%,单个最大杂质0.05%。结果见表4。
[0076]
表4所得酮咯酸氨丁三醇的hplc检测结果
[0077]
峰号保留时间面积面积%高度ntp(usp)分离度(usp)拖尾因子rrt18.07116160.0071268398
‑‑
1.10.29214.84929630.0123707136423.591.00.54316.854131550.0531142589098.021.80.61418.41648630.020320573575.340.70.66520.675121870.049962587016.961.00.75622.19812340.00582413663.92
‑‑
0.80722.86668020.027423494261.580.80.83825.52039840.016178282345.211.00.92927.7112473414899.7311233433432833.841.01.001034.75418280.00713513992815.581.01.251138.11281310.0333571458088.710.81.381239.16477600.0314881358602.551.01.411342.26721620.0091181141516.700.91.53总计 24800834100.0001238133
ꢀꢀꢀꢀ
[0078]
实施例2
[0079]
将m-2的甲苯、1,2-二氯乙烷混合溶液(以sm计,0.234mol,1.0equiv.),四丁基溴化铵3.76g(0.012mol,0.05equiv.),氢氧化钾水溶液约233ml(将46.7g氢氧化钾加入到233ml纯化水中进行溶解配置,纯化水预先进行脱氧)加入到反应器中,氩气保护。加热至35
~45℃反应完全,分液,水相用乙酸乙酯洗涤两次。氩气保护下,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,得到的m-3湿品直接加入甲基叔丁基醚打浆处理,过滤,真空干燥即可得到m-354.6g。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色2号和3号标准比色液之间。
[0080]
将m-3按照cn101143865进行制备,得到的酮咯酸氨丁三醇按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色0.5号和1号标准比色液之间,炽灼残渣0.06%,其纯度99.70%,单个最大杂质0.06%。
[0081]
实施例3
[0082]
将m-2的甲苯、1,2-二氯乙烷混合溶液(以sm计,0.234mol,1.0equiv.),四丁基氯化铵3.33g(0.012mol,0.05equiv.),氢氧化钠水溶液约233ml(将46.7g氢氧化钾加入到233ml纯化水中进行溶解配置,纯化水预先进行脱氧)加入到反应器中,氮气保护。加热至35~45℃反应完全,分液,水相用乙酸乙酯洗涤两次。氮气保护下,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,得到的m-3湿品直接加入异丙醚化合物打浆处理,过滤,真空干燥即可得到m-3 55.2g。按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色2号和3号标准比色液之间。
[0083]
将m-3按照cn101143865进行制备,得到的酮咯酸氨丁三醇按照中国药典溶液颜色检查法进行检查,其溶液颜色介于黄绿色0.5号和1号标准比色液之间,炽灼残渣0.04%,其纯度99.78%,单个最大杂质0.06%。
[0084]
下表是将对照例1-3和实施例1-3获得的酮咯酸(即干燥后的m-3)的颜色、纯度等检测结果汇总,见表5。
[0085]
表5:对照例1-3和实施例1-3的酮咯酸(m-3)的颜色检测结果
[0086][0087]
下表是将对照例1-3和实施例1-3获得的酮咯酸氨丁三醇(即成盐干燥后的样品)的颜色、炽灼残渣、纯度等检测结果汇总,见表6。
[0088]
表6:对照例1-3和实施例1-3的酮咯酸氨丁三醇的检测结果
[0089][0090]
从以上表的数据表明,本发明的方法不仅解决了现有技术中水解脱羧步骤产生的
颜色杂质难题,而且也避免了现有技术额外加入含硫或硼还原剂带来的成品中炽灼残渣较高的弊端。同时,直接在反应和后处理中全程进行惰性气体保护,操作简单、便捷,且在后处理中用醚类化合物替代醇和水纯化的繁琐步骤,有效地解决了酮咯酸氨丁三醇合成中颜色杂质带来的潜在困扰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1