一种咪唑并吡啶类染料及其合成方法和应用
1.本发明涉及有机荧光荧光染料合成领域,具体涉及具有高灵敏度、高选择性的咪唑并吡啶类染料及其合成方法和应用。
背景技术:
2.咪唑并吡啶母体结构是一类光学性能优异的荧光团,在其母体结构上进行修饰可得到不同吸收与发射波长的荧光化合物,表现出高摩尔吸光系数、高荧光量子产率、大斯托克斯位移和良好的光稳定性等优点,成为在荧光染料、生物成像、荧光探针等领域具有研究与应用价值的荧光分子。相比于传统技术,在水杨醛上引入供电子基形成一个“推
‑
拉”结构,扩大共轭体系、电子的离域和增加活性位点,从而调节其荧光性能,可以用来构造功能有机荧光染料分子,拓展了其在荧光染料上的应用。
技术实现要素:
3.针对现有技术存在的上述技术问题,本发明的目的在于提供一种咪唑并吡啶类染料及其合成方法和应用,本发明的咪唑并吡啶类染料的合成工艺相对简单、生产成本低廉、生产绿色环保。
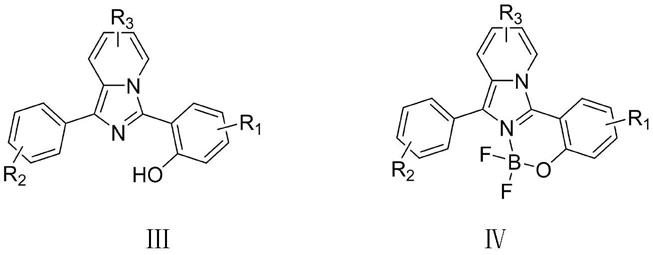
4.所述的一种咪唑并吡啶类染料,其特征在于其化学结构式如式(ⅲ)或(ⅳ)所示:
[0005][0006]
式(ⅲ)和(ⅳ)中,r1、r2、r3各自独立地选自h、羟基、硝基、卤素、氰基、氨基、c1~c6烷氧基、c1~c
16
的直链或支链烷基、取代的c1~c
16
的直链或支链烷基、取代芳基、单烷基取代的胺基或二烷基取代的胺基,所述单烷基取代的胺基或二烷基取代的胺基中的烷基的碳原子数为1~16个;
[0007]
所述取代的c1~c
16
的直链或支链烷基的取代基为一个或多个,各个取代基各自独立地选自c1~c
16
烷氧基、羟基、硝基或卤素;
[0008]
取代芳基的芳环上的取代基为一个或多个,各个取代基各自独立地选自c1~c
16
烷基、c1~c
16
烷氧基、羟基、硝基、卤素、胺基、单烷基取代的胺基或二烷基取代的胺基。
[0009]
所述的一种咪唑并吡啶类染料,其特征在于式(ⅲ)和(ⅳ)中,r2、r3均选自h,r1选自h、硝基、c1~c3烷氧基或二烷基取代的胺基,二烷基取代的胺基中的烷基的碳原子数为1~4个。
[0010]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于在催化剂存在作用下,式
(ⅰ)所示的2
‑
苯甲酰吡啶类化合物与式(ⅱ)所示的水杨醛类化合物反应生成式(ⅲ)所示的化合物;氮气保护下,且在碱存在条件下,式(ⅲ)所示的化合物与无水三氟化硼乙醚在溶剂中反应,得到相应的式(ⅳ)所示的n,o
‑
氟硼络合物,反应式如下:
[0011][0012]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于具体步骤如下:
[0013]
在催化剂存在作用下,式(ⅱ)所示的水杨醛类化合物与式(ⅰ)所示的2
‑
苯甲酰吡啶类化合物混合溶于乙酸中,回流条件下搅拌反应,反应结束后,反应液再经后处理得到式(ⅲ)所示的化合物;式(ⅲ)所示的化合物在氮气保护下,以二异丙基乙胺为碱,无水甲苯作为溶剂,90
‑
110℃下与无水三氟化硼乙醚反应,得到相应的式(ⅳ)所示的n,o
‑
氟硼络合物。
[0014]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于所述催化剂为乙酸胺;式(ⅰ)所示的2
‑
苯甲酰吡啶类化合物与所述催化剂的投料摩尔比为1:5~15,优选1:10~12;式(ⅱ)所示的化合物与式(ⅰ)所示的2
‑
苯甲酰吡啶类化合物的投料摩尔比为0.8~1.5:1,优选为1~1.1:1。
[0015]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于乙酸的体积与式(ⅰ)所示的2
‑
苯甲酰吡啶类化合物的物质的量之比为4~8:1,优选为5~6:1,体积的单位为ml,物质的量的单位为mmol。
[0016]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于回流状态下搅拌反应的时间为5~8h,优选为6~7h。
[0017]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于反应液经后处理的步骤为:向反应液中加蒸馏水,用二氯甲烷萃取,萃取相经无水硫酸钠干燥、浓缩除去溶剂后,进行柱层析分离得到式(ⅲ)所示的化合物。
[0018]
所述的一种咪唑并吡啶类染料的合成方法,其特征在于柱层析分离所用洗脱溶剂为乙酸乙酯与石油醚的混合液,乙酸乙酯与石油醚的体积比为1:3
‑
5。
[0019]
所述的一种咪唑并吡啶类染料作为荧光染料的应用。
[0020]
相对于现有技术,本发明取得的有益效果是:
[0021]
(1)本发明在2
‑
苯甲酰吡啶与水杨醛形成咪唑并吡啶的母体结构下,再在水杨醛
上引入供电子基形成一个“推
‑
拉”结构,扩大共轭体系、电子的离域和增加活性位点,从而调节其荧光性能,可以用来构造功能有机荧光染料分子,拓展了其在荧光染料上的应用。
[0022]
(2)本发明合成的咪唑并吡啶类染料通过式(ⅰ)所示的2
‑
苯甲酰吡啶类化合物和(ⅱ)所示的水杨醛类化合物在催化剂存在作用下反应,实现了咪唑并[1,5
‑
a]吡啶类染料的合成,合成工艺相对简单、生产成本低廉、生产绿色环保;所合成的咪唑并[1,5
‑
a]吡啶类染料无论在溶液中还是固体状态下都有着良好的荧光,且在溶剂中具有较大的斯托克斯位移,在质子性溶剂中高达213nm,并且咪唑并[1,5
‑
a]吡啶类染料可以用于检测cu
2+
,具有选择性好,灵敏度高等优点。
[0023]
综上,本发明所述功能分子咪唑并[1,5
‑
a]吡啶类染料的合成方法和应用具有制备方法环保、简单、高荧光量子产率的优点,可以应用到荧光染料等领域,同时还可检测cu
2+
,选择性好,灵敏度高。
附图说明
[0024]
图1a为实施例3制备的络合前体化合物2
‑
2c分别在toluene、dcm、chcl3、ea、thf、mecn、dmf、dmso、etoh和meoh有机溶剂中的紫外光谱图;
[0025]
图1b为实施例3制备的络合前体化合物2
‑
2c分别在toluene、dcm、chcl3、ea、thf、mecn、dmf、dmso、etoh和meoh有机溶剂中的荧光光谱图;
[0026]
图2a为实施例3制备的氟硼络合化合物2
‑
3c分别在toluene、dcm、chcl3、ea、thf、mecn、dmf、dmso、etoh和meoh有机溶剂中的紫外光谱图;
[0027]
图2b为实施例3制备的氟硼络合化合物2
‑
3c分别在toluene、dcm、chcl3、ea、thf、mecn、dmf、dmso、etoh和meoh有机溶剂中荧光发射光谱;
[0028]
图3a为不同取代基的络合前体化合物2
‑
2a~2
‑
2d在toluene溶剂中的紫外光谱图;
[0029]
图3b为不同取代基的络合前体化合物2
‑
2a~2
‑
2d在toluene溶剂中的荧光发射光谱;
[0030]
图4a为不同取代基的氟硼络合化合物2
‑
3a~2
‑
3d在toluene溶剂中的紫外光谱图;
[0031]
图4b为不同取代基的氟硼络合化合物2
‑
3a~2
‑
3d在toluene溶剂中的荧光发射光谱;
[0032]
图5a为络合前体化合物2
‑
2c在不同ph下的dmso
‑
水混合溶剂中的紫外光谱图;
[0033]
图5b为络合前体化合物2
‑
2c在不同ph下的dmso
‑
水混合溶剂中的荧光发射光谱;
[0034]
图6a为氟硼络合化合物2
‑
3c在不同ph下的dmso
‑
水混合溶剂中的紫外光谱图;
[0035]
图6b为氟硼络合化合物2
‑
3c在不同ph下的dmso
‑
水混合溶剂中的荧光发射光谱;
[0036]
图7为络合前体化合物2
‑
2a~2
‑
2d固体的荧光发射光谱;
[0037]
图8为氟硼络合化合物2
‑
2a~2
‑
2d固体的荧光发射光谱;
[0038]
图9为络合前体化合物2
‑
2c优化出的稳定结构图;
[0039]
图10为氟硼络合化合物2
‑
3c优化出的稳定结构图。
具体实施方式
[0040]
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
[0041]
实施例1:
[0042]
在50ml的反应瓶中依次加入2
‑
苯甲酰吡啶(2.0mmol,0.3661g)、水杨醛(3.0mmol,0.3662g)和乙酸铵(10.0mmol,0.7705g),塞紧瓶塞,注射10ml冰醋酸溶液,氮气氛围下,回流条件下反应5h。反应tlc监测至结束,加入100ml蒸馏水,二氯甲烷萃取3次(每次30ml),合并有机相,无水硫酸钠干燥,减压除去溶剂,用柱层析法(洗脱剂为:v乙酸乙酯/v石油醚=1:4)分离得到2
‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚(以下简称2
‑
2a),浅灰色固态产品(0.2804g,收率为49%),其化学结构式为:
[0043][0044]
对得到的2
‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚进行核磁谱图分析,结果如下:1hnmr(500mhz,cdcl3)δ12.01(s,1h),8.53(d,j=7.3hz,1h),7.90(d,j=8.3hz,3h),7.78(d,j=6.6hz,1h),7.49(t,j=7.7hz,2h),7.37(d,j=7.5hz,1h),7.34(d,j=7.2hz,1h),7.19(d,j=8.2hz,1h),7.02(t,j=8.1hz,1h),6.90
‑
6.84(m,1h),6.71(t,j=7.3hz,1h).
[0045]
在50ml的史莱克瓶中加入络合前体化合物2
‑
2a(0.5mmol),史莱克瓶中的气体抽真空用氮气置换3次,氮气保护下加入无水甲苯10ml,二异丙基乙胺0.5ml,100℃下搅拌约40min后再缓慢加入三氟化硼乙醚0.5ml,反应搅拌过夜。tlc监测反应结束后,冷却至室温,加入10ml水搅拌5min后,将体系用二氯甲烷萃取,合并有机层并用无水硫酸钠干燥,减压除去溶剂,得粗产物。粗产物通过柱层析进一步纯化得化合物2
‑
3a。黄色固态产品(0.0420g,收率为30%)其化学结构式为:
[0046][0047]
对得到的化合物2
‑
3a进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ8.64(d,j=6.7hz,1h),7.91(d,j=8.0hz,1h),7.78(d,j=7.1hz,2h),7.70(d,j=8.0hz,1h),7.55(t,j=7.3hz,2h),7.51(d,j=7.3hz,1h),7.47(t,j=7.9hz,1h),7.31(d,j=7.3hz,1h),7.11(t,j=7.7hz,1h),7.05
–
6.97(m,2h).
[0048]
实施例2:
[0049]
在50ml的反应瓶中依次加入2
‑
苯甲酰吡啶(2.0mmol,0.3661g)、4
‑
甲氧基水杨醛(3.0mmol,0.4562g)和乙酸铵(10.0mmol,0.7705g),塞紧瓶塞,注射10ml冰醋酸溶液,回流条件下反应5h。反应tlc监测至结束,加入100ml蒸馏水,二氯甲烷萃取3次(每次30ml),合并有机相,无水硫酸钠干燥,减压除去溶剂,用柱层析法(洗脱剂为:v乙酸乙酯/v石油醚=1:
4)分离得到4
‑
甲氧基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚(以下简称2
‑
2b),深黄色固态产品(0.3098g,收率为49%),其化学结构式为:
[0050][0051]
对实施例2得到的4
‑
甲氧基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ12.21(s,1h),8.44(d,j=7.4hz,1h),7.87(t,j=9.3hz,3h),7.68(d,j=8.7hz,1h),7.49(t,j=7.8hz,2h),7.33(t,j=7.4hz,1h),6.83(dd,j=9.1,6.1hz,1h),6.73(d,j=2.6hz,1h),6.66(t,j=6.3hz,1h),6.59(dd,j=8.7,2.6hz,1h),3.87(s,3h).
[0052]
在50ml的史莱克瓶中加入络合前体化合物2
‑
2b(0.5mmol),史莱克瓶中的气体抽真空用氮气置换3次,氮气保护下加入无水甲苯10ml,二异丙基乙胺0.5ml,100℃下搅拌约40min后再缓慢加入三氟化硼乙醚0.5ml,反应搅拌过夜。tlc监测反应结束后,冷却至室温,加入10ml水搅拌5min后,将体系用二氯甲烷萃取,合并有机层并用无水硫酸钠干燥,减压除去溶剂,得粗产物。粗产物通过柱层析进一步纯化得化合物2
‑
3b,深黄色固态产品(0.0983g,收率为54%)其化学结构式为:
[0053][0054]
对得到的化合物2
‑
3b进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ8.53(d,j=6.8hz,1h),7.81(d,j=8.9hz,1h),7.77(d,j=7.1hz,2h),7.64(d,j=8.3hz,1h),7.54(t,j=7.3hz,2h),7.51
–
7.46(m,1h),6.99
–
6.90(m,2h),6.84(d,j=2.5hz,1h),6.68(dd,j=8.8,2.6hz,1h),3.88(s,3h).
[0055]
实施例3:
[0056]
在50ml的反应瓶中依次加入2
‑
苯甲酰吡啶(2.0mmol,0.3661g)、4
‑
二乙氨基水杨醛(3.0mmol,0.5793g)和乙酸铵(10.0mmol,0.7705g),塞紧瓶塞,注射10ml冰醋酸溶液,回流条件下反应5h。反应tlc监测至结束,加入100ml蒸馏水,二氯甲烷萃取3次(每次30ml),合并有机相,无水硫酸钠干燥,减压除去溶剂,用柱层析法(洗脱剂为:v乙酸乙酯/v石油醚=1:4)分离得到4
‑
二乙氨基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚(以下简称2
‑
2c),浅黄色固态产品(0.4143g,收率为58%),其化学结构式为:
[0057][0058]
对实施例3得到的4
‑
二乙氨基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ12.06(s,1h),8.43(d,j=7.4hz,1h),7.89(d,j=7.1hz,2h),7.84(d,j=9.3hz,1h),7.62(d,j=8.7hz,1h),7.48(t,j=7.8hz,2h),7.31(t,j=7.4hz,1h),6.82
‑
6.73(m,1h),6.62(t,j=6.6hz,1h),6.48(s,1h),6.36(s,1h),3.41(q,j=7.0hz,4h),1.23(t,j=7.1hz,6h).
13
c nmr(126mhz,dmso
‑
d6)δ156.66(1c),149.35(1c),137.31(1c),134.94(1c),131.16(1c),128.76(1c),128.73(1c),126.18(1c),125.89(1c),125.77(1c),124.12(1c),124.02(1c),120.15(1c),118.18(1c),118.12(1c),112.17(1c),103.46(1c),103.29(1c),98.09(1c),43.82(2c),12.55(2c).
[0059]
在50ml的史莱克瓶中加入络合前体化合物2
‑
2c(0.5mmol),史莱克瓶中的气体抽真空用氮气置换3次,氮气保护下加入无水甲苯10ml,二异丙基乙胺0.5ml,100℃下搅拌约40min后再缓慢加入三氟化硼乙醚0.5ml,反应搅拌过夜。tlc监测反应结束后,冷却至室温,加入10ml水搅拌5min后,将体系用二氯甲烷萃取,合并有机层并用无水硫酸钠干燥,减压除去溶剂,得粗产物。粗产物通过柱层析进一步纯化得化合物2
‑
3c,浅黄色固态产品(0.1600g,收率为79%)其化学结构式为:
[0060][0061]
对得到的化合物2
‑
3c进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ8.42(d,j=7.3hz,1h),7.77(d,j=7.3hz,2h),7.68(d,j=9.0hz,1h),7.56(d,j=9.2hz,1h),7.52(t,j=7.4hz,2h),7.45(t,j=7.4hz,1h),6.87
–
6.83(m,1h),6.81(t,j=6.1hz,1h),6.53(d,j=2.4hz,1h),6.39(d,j=8.0hz,1h),3.41(q,j=7.1hz,4h),1.21(t,j=7.1hz,6h).
[0062]
实施例4:
[0063]
在50ml的反应瓶中依次加入2
‑
苯甲酰吡啶(2.0mmol,0.3661g)、5
‑
硝基水杨醛(3.0mmol,0.5010g)和乙酸铵(10.0mmol,0.7705g),塞紧瓶塞,注射10ml冰醋酸溶液,回流条件下反应5h。反应tlc监测至结束,加入100ml蒸馏水,二氯甲烷萃取3次(每次30ml),合并有机相,无水硫酸钠干燥,减压除去溶剂,用柱层析法(洗脱剂为:v乙酸乙酯/v石油醚=1:4)分离得到5
‑
硝基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚(以下简称2
‑
2d),黄色固态产品(0.5099g,收率为77%),其化学结构式为:
[0064][0065]
对得到的5
‑
硝基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ8.79(s,1h),8.59(d,j=7.3hz,1h),8.22(d,j=6.5hz,1h),7.98(d,j=9.2hz,1h),7.87(d,j=7.1hz,2h),7.52(t,j=7.8hz,2h),7.39(t,j=7.4hz,1h),7.22(d,j=9.1hz,1h),7.02(dd,j=8.9,6.1hz,1h),6.92(t,j=6.3hz,1h).
[0066]
在50ml的史莱克瓶中加入络合前体化合物2
‑
2d(0.5mmol),史莱克瓶中的气体抽真空用氮气置换3次,氮气保护下加入无水甲苯10ml,二异丙基乙胺0.5ml,100℃下搅拌约40min后再缓慢加入三氟化硼乙醚0.5ml,反应搅拌过夜。tlc监测反应结束后,冷却至室温,加入10ml水搅拌5min后,将体系用二氯甲烷萃取,合并有机层并用无水硫酸钠干燥,减压除去溶剂,得粗产物。粗产物通过柱层析进一步纯化得化合物2
‑
3d,黄色固态产品(0.1156g,收率为61%)其化学结构式为:
[0067][0068]
对得到的化合物2
‑
3d进行核磁谱图分析,结果如下:1h nmr(500mhz,cdcl3)δ8.91(d,j=2.6hz,1h),8.71(d,j=7.2hz,1h),8.36(dd,j=9.1,2.6hz,1h),7.80
–
7.74(m,3h),7.60
–
7.53(m,3h),7.38(d,j=9.1hz,1h),7.24
–
7.16(m,2h).
[0069]
实施例5:
[0070]
对于氟硼络合前体化合物2
‑
2在不同溶剂中荧光性能的研究,选择化合物2
‑
2c为例,准确称取本发明实施例3的化合物2
‑
2c加入到二氯甲烷中溶解配成10ml的溶液,配制得到化合物2
‑
2c的有效浓度为1
×
10
‑3mol
·
l
‑1的母液。取母液0.1ml,分别加入到相应的10ml容量瓶中,将二氯甲烷溶剂吹干,再分别加入10种溶剂配成浓度为1
×
10
‑5mol
·
l
‑1的4
‑
二乙氨基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚溶液备用。10种溶剂包括甲苯(toluene)、二氯甲烷(dcm)、氯仿(chcl3)、乙酸乙酯(ea)、四氢呋喃(thf)、乙腈(mecn)、n,n
‑
二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、乙醇(etoh)、甲醇(meoh)。
[0071]
于10mm的比色皿中分别加入上述配制的1
×
10
‑5mol
·
l
‑1的4
‑
二乙氨基
‑2‑
(1
‑
苯基咪唑并[1,5
‑
a]吡啶
‑3‑
基)苯酚溶液3ml,室温下分别测试其吸收光谱,并以最大吸收波长作为激发波长进行发射光谱的测试,以积分球法测量绝对量子产率。其结果分别如表1、图1a、图1b所示。
[0072]
表1
[0073][0074]
化合物2
‑
2c在不同溶剂中的吸收和发射光谱如表1和图1a所示。由图1b中可以看到化合物2
‑
2c的荧光发射波长约为520nm,其溶液的荧光颜色为黄绿色。在不同溶剂中,荧光发射峰峰形基本一致,但强度略有差别。在toluene、thf、dmso中荧光量子产率相对较高,在dmso中最高可达到0.17,但在质子性溶剂meoh、etoh中较低,仅为0.05左右。化合物2
‑
2c在各种不同极性溶剂中都具有较大的斯托克斯位移(≥170nm),且随溶剂极性的增加而增大,在强极性溶剂dmf、dmso和质子性溶剂meoh、etoh中可达到200nm以上。在其荧光发射光谱中并没有观察到化合物2
‑
2c因esipt效应所导致的双重发射,推测酚羟基的o原子与咪唑并[1,5
‑
a]吡啶环上的n原子没有发生激发态分子内质子转移。
[0075]
实施例6:
[0076]
对于氟硼络合化合物2
‑
3在不同溶剂中荧光性能的研究,将化合物2
‑
3c分别溶解在10种不同的溶剂中,化合物2
‑
3c在溶剂中的有效浓度均为1
×
10
‑5mol
·
l
‑1。将化合物2
‑
3c分别溶解在10种不同的溶剂中,对其紫外可见吸收光谱和荧光发射光谱进行了测试,测试方法重复实施例5,结果如表2和图2a、图2b所示。
[0077]
从表2和图2a、图2b中可以看出,化合物2
‑
3c在除了质子性溶剂meoh、etoh外的其余几种溶剂中,其紫外可见吸收峰峰形基本一致,最大吸收峰约位于370nm处,吸收强度也并无太大差异,摩尔消光系数在3.0
×
104m
‑1cm
‑1上下。而在meoh、etoh中吸收波长蓝移至303nm,吸收强度明显降低,摩尔消光系数降至2.4
×
104m
‑1cm
‑1。在荧光发射光谱方面,其在非极性溶剂中发射波长在510nm左右,而在极性溶剂中其红移了5~10nm,在质子性溶剂meoh、etoh中其荧光强度明显降低,荧光量子产率由toluene中最高的0.16降至0.04,斯托克斯位移却达到了217nm。而化合物2
‑
3c在其余8种溶剂中的荧光强度差异不大,斯托克斯位移在130~150nm范围内。通过分析化合物2
‑
3c在meoh、etoh中的紫外吸收和荧光发射的变化,发现其与络合前体化合物2
‑
2c在meoh、etoh中的紫外吸收波长、摩尔消光系数以及荧光发射波长和荧光量子产率等参数基本吻合,经tlc监测分析,猜想氟硼络合化合物2
‑
3c在质子性溶剂中不稳定,可能分解为化合物2
‑
2c。
[0078]
表2
[0079][0080]
实施例7:
[0081]
探究取代基对络合前体化合物荧光性能的影响,选择toluene作为溶剂,对化合物2
‑
2a~2
‑
2d进行了取代基效应的测试。
[0082]
将化合物2
‑
2a、化合物2
‑
2b、化合物2
‑
2c和化合物2
‑
2d分别溶解于toluene溶剂中,均配制化合物有效浓度为1
×
10
‑5mol
·
l
‑1的溶液,测试其紫外可见吸收光谱和荧光发射光谱,测试方法重复实施例5,结果如表3和图3a、图3b所示。
[0083]
表3
[0084][0085]
从图3a、图3b中可以看出,化合物2
‑
2a和2
‑
2b的最大紫外可见吸收波长都在340nm左右,摩尔消光系数差异不大。化合物2
‑
2b由于甲氧基的给电子效应,发射波长较2
‑
2a红移了12nm,但都呈蓝色荧光。当4号位引入较强推电子基二乙氨基时,化合物2
‑
2c的吸收波长和发射波长都较化合物2
‑
2a发生了红移,摩尔消光系数达到2.5
×
104m
‑1·
cm
‑1,呈最大荧光强度发射波长523nm的绿色荧光。然而,强吸电子基硝基的引入,使化合物2
‑
2d的最大荧光强度发射波长高达576nm,在实现长波长发射的同时却大大降低了其荧光强度(ф
f
≤0.01),因为与给电子基相比,其n
→
π*的跃迁是禁阻的,s1‑
t1的系间跨越占优势,其所释放的激发态分子数减少,致使荧光减弱。
[0086]
几个化合物都具有较大的斯托克斯位移,其中化合物2
‑
2d在toluene中的斯托克斯位移可以达到215nm,化合物2
‑
2a、2
‑
2b、2
‑
2c也都在140nm以上。这类大斯托克斯位移的荧光分子具有背景干扰低、样品穿透性强、检测灵敏度高等特点,可用在生物成像、荧光传感器等方面。
[0087]
实施例8:
[0088]
探究取代基对氟硼络合化合物2
‑
3a~2
‑
3d荧光性能的影响,选择toluene作为溶剂,对化合物2
‑
3a~2
‑
3d进行了取代基效应的测试。
[0089]
将化合物2
‑
3a、化合物2
‑
3b、化合物2
‑
3c和化合物2
‑
3d分别溶解于toluene溶剂中,均配制化合物有效浓度为1
×
10
‑5mol
·
l
‑1的溶液,测试其紫外可见吸收光谱和荧光发射光谱,测试方法重复实施例5,结果如表4和图4a、图4b所示。
[0090]
表4
[0091][0092]
从图4a、图4b中可以看出,化合物2
‑
3a与2
‑
3b的最大紫外可见吸收波长都在355nm左右,吸收强度也相差不大,摩尔消光系数约为1.7
×
104m
‑1·
cm
‑1。但甲氧基作为推电子基,使化合物2
‑
3b相对于化合物2
‑
3a的发射波长由456nm红移至472nm。而强推电子基二乙氨基的引入使化合物2
‑
3c的紫外可见吸收波长和荧光发射波长都发生了红移,分别为375nm的最强吸收波长和最大荧光强度发射波长508nm的蓝绿色荧光,摩尔消光系数也增加至2.9
×
104m
‑1·
cm
‑1。化合物2
‑
3d由于硝基的强吸电子效应虽使得其荧光发射波长相对于化合物2
‑
3a发生了红移,但其荧光量子产率≤0.01,荧光几乎淬灭,而2
‑
3a~2
‑
3c的荧光量子产率在0.16~0.24之间。这四个化合物的斯托克斯位移也都在100nm以上,这在bodipy衍生物中也是很少见的。
[0093]
实施例9:
[0094]
探究不同ph对络合前体化合物2
‑
2c荧光性能影响,将化合物2
‑
2c溶解在dmso
‑
水混合溶剂(dmso/h2o,9:1,v/v)中,将化合物2
‑
2c配制成有效浓度为1.0
×
10
‑5mol l
‑1的溶液,加入稀盐酸或者氢氧化钠溶液调节体系的ph为2至13,然后分别测试其在不同ph条件下的紫外可见吸收光谱和荧光发射光谱,结果如图5a、5b所示。
[0095]
从图5a、5b中可以看出,化合物2
‑
2c在强酸性条件下的紫外可见吸收和荧光发射光谱有着明显的差异。当溶液的ph≤5时,其吸收峰的峰形和强度变化较为明显。随着酸性增强,吸收强度略有下降,在290nm与335nm处出现了两个新的吸收峰。当ph>5时,随着碱性的增强,其吸收峰峰形和强度没有变化,吸收主峰位于313nm处,与化合物2
‑
2c在纯dmso溶剂中一致。而在荧光发射光谱中可以看到,当溶液的ph≤5时,随着酸性的增强,在400nm处出现了强度较低的新发射峰,最大发射波长蓝移至505nm处,且荧光强度大大增强,这归因于咪唑环上n原子的质子化抑制了激发态的分裂,在质子化过程中,孤对电子处于激发态,较难跃迁回基态,故在ph≤5时发生轻微的蓝移,其可作为极酸性环境下的荧光传感器。当ph>5时,其发射波长在520nm左右且强度差异不大。在加入一定量的氢氧化钠或盐酸溶液后,其吸收和发射峰都会恢复,对ph的检测具有很好的可逆性。
[0096]
实施例10:
[0097]
探究不同ph对氟硼络合化合物2
‑
3c荧光性能影响,将化合物2
‑
3c溶解在dmso
‑
水混合溶剂(dmso/h2o,9:1,v/v)中,将化合物2
‑
2c配制成有效浓度为1.0
×
10
‑5mol l
‑1的溶液,加入稀盐酸或者氢氧化钠溶液调节体系的ph为2至13,然后分别测试其在不同ph条件下的紫外可见吸收光谱和荧光发射光谱,结果如图6a、6b所示。
[0098]
从图6a、6b中可以看出,化合物2
‑
3c在ph=2~4的强酸性范围内,其紫外可见吸收峰略有变化。当ph≤4时,吸收峰变宽,其最大吸收波长相对于中性或碱性发生了蓝移,吸收强度明显降低,摩尔消光系数降至约1.4
×
104m
‑1·
cm
‑1。当体系ph>4时,其吸收波长在370nm左右,摩尔消光系数基本不变,其峰形和波长都与化合物2
‑
3c在纯dmso溶剂中一致。在荧光发射光谱中可以看到,ph为2或3时,其发射峰形有所改变,位于约420nm处的位置出现了逐渐增强的拐点,最大发射波长也发生了轻微的蓝移,由520nm的常规发射蓝移至515nm,但其在不同ph条件下的荧光颜色和强度差异不大。
[0099]
实施例11:
[0100]
探究络合前体目标化合物2
‑
2a~2
‑
2d的固体荧光性能,取代基的引入使得化合物2
‑
2a~2
‑
2d呈现出不同的固体荧光,因此对该类化合物进行了固体荧光性能的检测,其结果如表5和图7所示。
[0101]
从图7的荧光发射光谱中可以看到,化合物2
‑
2a~2
‑
2d固体状态下的荧光发射波长都在490nm以上,相对于其在溶液中发生了略微的红移,这是分子平面性和分子间相互作用影响的结果。化合物2
‑
2a的固体荧光量子产率达到了0.17,具有蓝绿色的荧光。取代基的引入会导致荧光分子固体的量子产率降低,可能是分子的堆积方式或者分子平面性的改变所致。硝基的引入虽然使化合物在溶液中的荧光几乎淬灭,但其固体却显示出最大荧光强度发射波长约在591nm的黄色固体荧光,且斯托克斯位移为111nm,在几个化合物中也是最大的。
[0102]
表5
[0103][0104]
实施例12:
[0105]
探究氟硼络合化合物2
‑
3a~2
‑
3d的荧光性能,取代基的引入也使得化合物2
‑
3a~2
‑
3d呈现出不同的固体荧光,因此对该类化合物也进行了固体荧光性能的检测,其结果如表6和图8所示。
[0106]
从图8中可以看到,化合物2
‑
3a~2
‑
3c都为浅黄色固体粉末,在固体状态下显现出最大荧光强度发射波长分别为465nm、491nm和485nm的蓝绿色固体荧光。而硝基取代的化合物2
‑
3d则为橙黄色固体粉末,与其在溶液中的荧光性质类似,其固体荧光也处于淬灭状态,可能是由于氟硼络合所带来的高平面性原因所致。在这种情况下,分子聚集后造成非常强烈的分子间相互作用,进而导致荧光淬灭,即发生了acq效应。化合物2
‑
3a~2
‑
3c虽具有固体荧光,但其固体的荧光量子产率都很低,均小于0.05。总的来说,甲氧基与二乙氨基等推
电子基的引入使化合物2
‑
3b与2
‑
3c的固体荧光发射波长相对于化合物2
‑
3a发生了较为明显的红移,即使硝基作为强吸电子基,其发射波长也高达602nm。因此无论供电子基还是吸电子基的引入都会增强分子内电荷转移(ict),但较强的ict效应所带来的非辐射能增加也是导致发射波长红移与荧光量子产率降低的主要因素。
[0107]
表6
[0108][0109]
实施例13:
[0110]
探究络合前体化合物2
‑
2a~2
‑
2d光谱性质的理论计算,化合物2
‑
2c优化出的稳定结构如图9所示,咪唑并[1,5
‑
a]吡啶杂环两侧的取代苯环都具有一定角度的旋转,这种适宜的旋转角也为金属离子的配位提供了非常好的空间。另外,采用酮式结构输入,优化的输出结果同样为烯醇式,因此推测化合物2
‑
2c以烯醇式的稳定结构存在。
[0111]
表7为化合物2
‑
2a~2
‑
2d在td
‑
dft方法下计算出的光学数据,可以看到理论计算的紫外可见吸收波长、荧光发射波长与实际测得的数据几乎一致,很好地验证了实验数据的准确性。从能级水平来看,化合物2
‑
2c与2
‑
2d的能级差分别为4.05ev、4.02ev,低于化合物2
‑
2a与2
‑
2b,因此发生电子跃迁时所需的能量较小,故化合物2
‑
2c与2
‑
2d的吸收和发射波长相对于2
‑
2a与2
‑
2b都发生了较大的红移。
[0112]
表7
[0113][0114]
实施例14:
[0115]
探究氟硼络合化合物2
‑
3a~2
‑
3d光谱性质的理论计算,化合物2
‑
3c优化出的稳定结构如图10所示,咪唑并[1,5
‑
a]吡啶杂环与取代的苯酚环被bf2结构固定,限制了单键的旋转,n
‑
b键的键长为o
‑
b键的键长为由于键长的差异,氟硼六元环的平面出现了轻微的扭曲,但整体平面性良好。
[0116]
表8为化合物2
‑
3a~2
‑
3d在td
‑
dft方法下计算出的光学数据,可以看到理论计算的紫外可见吸收波长与实际测得的数据差别不大,其荧光发射波长则具有相同的趋势,理论计算很好地验证了实验数据的准确性。另外,化合物2
‑
3c与2
‑
3d的能级差也小于2
‑
3a与
2
‑
3b,对应为吸收和发射波长的红移。通过电子跃迁和理论计算的光谱数据可以看到,因其跃迁方式和电子云密度分布都类似,因此氟硼络合化合物2
‑
3a~2
‑
3d与络合前体化合物2
‑
2a~2
‑
2d相比并没有表现出太大的差异性,这与之前的测试结果相符。
[0117]
表8
[0118][0119]
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1