一种活性染料化合物及其制备方法和应用与流程
一种活性染料化合物及其制备方法和应用
(一)技术领域
1.本发明涉及一种活性染料化合物,特别涉及一种活性染料化合物及其制备方法和在纤维素纤维、聚酰胺纤维及其织物印染中的应用。
(二)
背景技术:
2.活性染料上染织物的耐水洗牢度的提高,一直是印染生产中的重点和难点。部分活性染料与棉纤维间的共价键受外界条件的影响,易发生断键水解,从而引起染色织物的色牢度变差,特别是耐水洗沾色牢度不能很好地满足生产要求。为解决上述问题,本发明人开发了新的活性染料结构,采用3,5
‑
二羟基苯甲酸为偶合组分,并对该化合物进行了深入细致的研究,在大量实验的基础上,获得了各项色牢度性能俱佳的黄色至橙色染料。
(三)
技术实现要素:
3.本发明的目的在于提供结构新颖、性能优异的活性染料化合物及其制备方法和在纤维素纤维、聚酰胺纤维及其织物印染中的应用,该染料各项色牢度优异,特别是耐水洗牢度和耐摩擦牢度突出,与纤维结合稳定性好,能满足棉、人造棉、丝绸、粘胶、羊毛及其混纺织物的常规染色和印花的要求。
4.下面对本发明所采用的技术方案进行具体说明。
5.第一方面,本发明提供了一种活性染料化合物,其为结构如下式(ⅰ)所示的化合物或其碱金属盐:
[0006][0007]
式(ⅰ)中:
[0008]
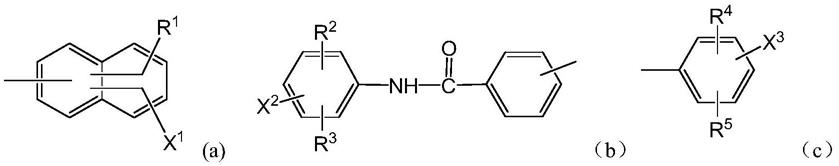
d1、d2各自独立为下式(a)、式(b)或式(c)式所示的基团,且d1、d2不同时为下式(a)、式(b) 或式(c)所示的基团:
[0009][0010]
上式(a)~(c)中:
[0011]
r1~r5各自独立为h、c1~c4直链或支链烷基、c1~c4烷氧基或磺酸基;
[0012]
x1、x2、x3彼此独立为h、c1~c4直链或支链烷基、c1~c4烷氧基、
‑
so2y1、
‑
nhco(ch2)
p
so2y2或
ꢀ‑
conh(ch2)
q
so2y3,且d1、d2中至少有一个含有纤维活性基团,即
‑
so2y1、
‑
nhco
(ch2)
p
so2y2或
‑
conh(ch2)
q
so2y3,其中y1~y3各自独立为
‑
ch=ch2、
‑
c2h4oso3h或
‑
ch2ch2cl,p=1
‑
3,q=1
‑
3。
[0013]
本发明中,所述的c1~c4直链或支链烷基可以是甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基等;所述的c1~c4烷氧基可以是甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基等;所述的c1~c4的烷酰氨基可以是甲酰氨基、乙酰氨基、正丙酰氨基、异丙酰氨基、正丁酰氨基、异丁酰氨基、叔丁酰氨基等。
[0014]
进一步,式(a)~(c)中:
[0015]
r1~r5各自独立为h、甲基、甲氧基或磺酸基;
[0016]
x1、x2、x3彼此独立为
‑
so2y1、
‑
nhco(ch2)
p
so2y2或
‑
conh(ch2)
q
so2y3,其中y1~y3各自独立为
ꢀ‑
ch=ch2、
‑
ch2ch2cl或
‑
c2h4oso3h,p=1
‑
3,q=1
‑
3。
[0017]
进一步,d1优选式(a)或式(b)所示的基团,d2优选式(c)所示的基团,所述的活性染料化合物具有下式(ⅰa)或式(ⅰb)所示的结构:
[0018][0019]
式(ⅰa)、式(ⅰb)中各取代基定义同式(ⅰ)。
[0020]
更进一步,所述的活性染料化合物中,所述的式(a)优选自下列基团之一:
[0021]
[0022]
更进一步,所述的活性染料化合物中,所述的式(b)优选自下列基团之一:
[0023][0023][0024]
更进一步,所述的活性染料化合物中,所述的式(c)优选自下列基团之一:
[0025][0026]
本发明具体推荐所述活性染料化合物选自下列式(i
‑
1)~(i
‑
63)所示的化合物及其碱金属盐中的一种:
[0027]
[0028]
[0029]
[0030]
[0031]
[0032]
[0033]
[0034]
[0035]
[0036]
[0037]
[0038]
[0039]
[0040][0041]
进一步,本发明优选所述活性染料化合物选自式(
ⅰ‑
1)~(
ⅰ‑
40)所示的化合物及其碱金属盐中的 一种。
[0042]
更进一步,本发明优选所述活性染料化合物选自式(
ⅰ‑
1)~(
ⅰ‑
8)、式(
ⅰ‑
11)~(
ⅰ‑
26)所示的化合物及其碱金属盐中的一种。
[0043]
需要强调的是,本发明所述的染料化合物(ⅰ),在酚羟基邻位偶合的染料经常以醌腙体形式存在,亦即通式(ⅰ)的染料混合物含有下式(ⅰe)和/或(ⅰf)和/或(ⅰg)和/或(ⅰh)所示的醌腙体结构,考虑到本领域技术人员的书写习惯,在发明内容和实施例部分仍采用了偶氮体的书写形式,这并不影响本发明的实质:
[0044][0045]
式(ⅰe)~(ⅰh)中各取代基定义同式(ⅰ)。
[0046]
第二方面,本发明提供了一种式(ⅰ)所示的化合物的制备方法,包括:
[0047]
(1)重氮化:
[0048]
根据需要将式(ⅱa)、式(ⅱb)和/或式(ⅱc)所示的芳胺化合物分别进行重氮化,得到各自的重氮盐;
[0049]
(2)偶合反应:
[0050]
将3,5
‑
二羟基苯甲酸加水打浆,打浆好的溶液与步骤(1)中的式(ⅱa)的重氮盐、式(ⅱb)的重氮盐、式(ⅱc)的重氮盐中的其中之一进行一次偶合,然后再与式(ⅱa)的重氮盐、式(ⅱb)的重氮盐、式(ⅱc)的重氮盐的其中之一进行二次偶合,并且,一次偶合和二次偶合所采用的重氮盐不能均为式(
ⅱꢀ
a)的重氮盐、式(ⅱb)的重氮盐或式(ⅱc)的重氮盐,即得到所述的化合物(ⅰ);
[0051][0052]
上述式(ⅱa)~式(ⅱc)中各取代基定义同式(ⅰ)。
[0053]
进一步,所述化合物(ⅰ)优选合成步骤如下:
[0054]
(1)重氮化:
[0055]
根据需要将式(ⅱa)、式(ⅱb)和/或式(ⅱc)所示的化合物各自与水、冰一起打浆1~2h,打浆完成后加入一定量的盐酸,然后在20
‑
30min内加入亚硝酸钠溶液,控制ph=0.5
‑
3.0(优选为0.5
‑
2.0)、温度 t=0
‑
30℃(优选为0
‑
20℃)进行重氮化反应,用4
‑
二甲氨基苯甲醛的乙醇溶液检测终点(即5s内不变色);重氮终点到后,用氨基磺酸消除过量的亚硝酸钠,分别得到式(ⅱa)、式(ⅱb)或式(ⅱc)重氮液保存备用;其中,式(ⅱa)、式(ⅱb)或式(ⅱc)化合物各自与盐酸、亚硝酸钠的摩尔比为1:(1
‑
3):(1
‑
1.1),优选为1:(1
‑
1.8):(1
‑
1.02);
[0056]
(2)偶合反应:
[0057]
将3,5
‑
二羟基苯甲酸加水打浆分散,控制温度为15
‑
25℃。将分散好的3,5
‑
二羟基苯甲酸加入上述步骤 (1)制得的式(ⅱa)的重氮液、式(ⅱb)的重氮液或式(ⅱc)的重氮液中,用液碱或小苏打控制ph 在2.0
‑
5.0、温度控制为0
‑
30℃(优选为0
‑
20℃)进行偶合反应,用h酸试液测试重氮,渗圈处无色即重氮已反应完全到终点,得到偶合产物1;
[0058]
再将步骤(1)得到的式(ⅱa)的重氮液、式(ⅱb)的重氮液或式(ⅱc)的重氮液加入到偶合产物 1中,用液碱或小苏打控制ph在5.0
‑
8.0、温度控制为0
‑
30℃(优选为0
‑
20℃)进行偶合反应,用h酸试液测试重氮,渗圈处无色即重氮已反应完全到终点,即可得到本发明所述的式(ⅰ)的化合物;
[0059]
上述两次偶合反应中所使用的重氮液不能均为式(ⅱa)的重氮液、式(ⅱb)的重氮液或式(ⅱc)的重氮液;其中,式(ⅱa)、式(ⅱb)、式(ⅱc)化合物与3,5
‑
二羟基苯甲酸的摩尔比为(0.95
‑
1.2):1,优选(0.98
‑
1.08):1。
[0060]
本领域公知,本发明式(ⅰ)的化合物中,当y1~y3为
‑
c2h4oso3h或
‑
ch2ch2cl时,在通常的活性染料染色应用中,β
‑
羟乙砜基硫酸酯(
‑
so2c2h4oso3h)或β
‑
氯乙基砜(
‑
so2c2h4cl)在碱性介质中经消除反应生成乙烯砜基(
‑
so2ch=ch2),然后与纤维素纤维经亲核加成反应,形成共价键。
[0061]
本发明所述的化合物(ⅰ),在其制备过程中,还允许存在含量不超过30%的副产
物,所述的副产物可不分离,直接用于商品染料的加工,包括且不限于以下结构的化合物或混合物:
[0062][0063]
上述副产物中,各取代基定义同前述式(ⅰ)。
[0064]
本发明所述的活性染料化合物,式(i)体现为自由酸的形式,在实际合成过程中,通常以其碱金属盐 (优选钠盐或钾盐,尤其优选钠盐)的形式进行制备和分离,并且也以其盐的形式用于染色,这也是本领域技术人员所熟知的,亦即式(ⅰ)中的羧基、磺酸基可以羧酸钠、磺酸钠的形式存在。
[0065]
本发明所述的活性染料化合物可以粉状、粒状、水溶液或合成溶液形式存在,从合成溶液分离本发明所述的染料化合物,可用普遍已知的方法进行,例如用电解质(如氯化钠或氯化钾)从反应介质中将染料盐析滤出,或将溶液蒸发、喷雾干燥,因此,染料化合物通常包含有活性染料中常规的电解质盐(如氯化钠、硫酸钠等)。
[0066]
第三方面,本发明所述的活性染料化合物,作为商品出售时,可以不加入助剂,也可以加入商品染料中的常规助剂,如助溶剂、分散剂、耐碱性助剂、防尘剂、表面活性剂、缓冲剂和促染剂等。因此,本发明还提供了一种活性染料制品,其含有所述的活性染料化合物。优选的,所述的活性染料制品中,含有所述的活性染料化合物以及助剂,其中助剂的重量不超过活性染料化合物重量的45%,优选不超过40%。所述的助剂优选下列一种或任意几种的组合:萘磺酸甲醛缩合物(nno)、甲基萘磺酸甲醛缩合物(分散剂 mf)、扩散剂cnf(苄基萘磺酸盐甲醛缩合物)、元明粉(工业硫酸钠)、木质素磺酸盐、醋酸钠、碳酸氢钠、柠檬酸钠、磷酸二氢钠、磷酸氢二钠、增稠剂等。所述助剂均为市售常规品种。
[0067]
第四方面,本发明提供了所述的活性染料制品在纤维素纤维、聚酰胺纤维或其织物的印染中的应用。其中,纤维素纤维优选棉纤维或再生纤维,当然也可包括其它的植物性纤维,如麻类纤维或织物;聚酰胺纤维优选包括皮、毛或丝在内的动物性纤维材料,以及合成的尼龙6、尼龙66等纤维材料。采用本发明所述的活性染料化合物或活性染料制品印染上述纤维材料时,可遵照已知的活性染料染色方法进行,如常用的活性染料浸染染色法和轧染染色法,所述的浸染染色是将织物浸渍于染液中,使染料逐渐上染织物的方法,通常需经染色-固色-水洗-皂煮-水洗-脱水-烘干等工序。
[0068]
所述的轧染则是先把织物浸渍于染液中,然后使织物通过轧辊,把染液均匀轧入织物内部,再经汽蒸或热熔等处理的染色方法,通常需经浸轧染液-烘干-(浸轧固色液)-汽蒸或焙烘-水洗-皂洗-水洗-烘干等工序。
[0069]
通常,由于织物上染色泽要求的不同,染料使用量也会有所不同,使用浸染法染色时,染色深度(owf) 一般为0.1%~10%(染料占织物重量百分比),浴比1:2~1:60(织物与染液重量比,优选1:10~1:30),初染温度控制30~60℃,染色时间10~30分钟,皂煮温度85~95℃,皂煮时间10~15分钟,固色温度60~100℃,固色时间10~50分钟,固色ph值9~11。
使用轧染方法染色时,纤维素纤维的轧余率一般为60~80%,汽蒸温度100~103℃,汽蒸时间1~3分钟。轧染方法中,目前较多使用的是冷轧堆染色法,将染料和碱性物质引入到轧染机中,并在室温下打卷堆置2~30小时进行固色,之后进行彻底漂洗。
[0070]
本发明的有益效果在于:本发明的活性染料化合物结构新颖,各项色牢度优异,特别是耐水洗牢度和耐摩擦牢度突出,与纤维结合稳定性好,适用于棉、人造棉、丝绸、羊毛等纤维的染色和印花。
(四)附图说明
[0071]
图1是实施例3制备的活性染料化合物的紫外
‑
可见光谱图;
[0072]
图2是实施例3制备的活性染料化合物的质谱图,m/z=432.96为目标分子双电荷电离峰。。
(五)具体实施方式
[0073]
下面结合具体实施例对本发明作进一步描述,但本发明的保护范围并不仅限于此:
[0074]
本发明实施例中制备得到的染料化合物为钠盐,并以钠盐的形式进行应用,但为方便撰写,实施例中的所有结构式皆以自由酸的形式体现,本领域技术人员公知自由酸和钠盐的实质染色性能等同。
[0075]
实施例1
[0076]
(1)重氮化:
[0077]
将41.1g(0.1mol)2
‑
氨基
‑6‑
羟乙基砜硫酸酯基
‑1‑
萘磺酸投入100g水、100g冰中打浆约1h,再加20g 31%盐酸(含hcl 0.17mol),在20
‑
30min内加入24g 30%亚硝酸钠液(含亚硝酸钠0.104mol),控制ph=0.5
‑
2.0、温度t=0
‑
20℃进行重氮化反应1~2小时,用4
‑
二甲氨基苯甲醛的乙醇溶液检测终点(即5s内不变色)。重氮终点到后,用氨基磺酸消除过量的亚硝酸钠,得到的2
‑
氨基
‑6‑
羟乙基砜硫酸酯基
‑1‑
萘磺酸重氮液保存备用。
[0078]
将36.1g(0.1mol)磺化对位酯(2
‑
磺酸
‑4‑
β
‑
羟乙砜硫酸酯苯胺)投入100g水、100g冰中打浆约1h,再加20g 31%盐酸(含hcl 0.17mol),在20
‑
30min内加入24g 30%亚硝酸钠液(含亚硝酸钠0.104mol),控制ph=0.5
‑
2.0、温度t=0
‑
20℃进行重氮化反应1~2小时,用4
‑
二甲氨基苯甲醛的乙醇溶液检测终点(即 5s内不变色)。重氮终点到后,用氨基磺酸消除过量的亚硝酸钠,得到的磺化对位酯重氮液保存备用。
[0079]
(2)偶合:
[0080]
首先将154g(0.1mol)3,5
‑
二羟基苯甲酸加入200g水中进行打浆,控制温度20
‑
25℃,将打浆好的3,5
‑ꢀ
二羟基苯甲酸溶液加入到步骤(1)中得到的2
‑
氨基
‑6‑
羟乙基砜硫酸酯基
‑1‑
萘磺酸重氮液中,用30%液碱控制ph在2.0
‑
4.0,温度控制在0
‑
20℃,反应15
‑
20h,用hplc控制游离3,5
‑
二羟基苯甲酸在3%以下即到终点,得下式(a)所示的色基1。
[0081]
[0082]
将步骤(1)中制得的磺化对位酯重氮液加入到色基1中,用30%液碱控制ph在5.0
‑
8.0,温度t控制在0
‑
20℃,进一步偶合反应5
‑
10h,用h酸试液测试重氮,渗圈处无色即重氮反应完全到终点,得到橙色活性染料化合物(
ⅰ‑
1),水溶液中λmax=440nm。
[0083][0084]
实施例2
[0085]
(1)重氮化:
[0086]
将40.0g(0.1mol)4
‑
乙酰(4'
‑
β
‑
羟乙基砜基硫酸酯苯胺)基苯胺投入100g水、100g冰中打浆约1h,再加20g 31%盐酸(含hcl 0.17mol),在20
‑
30min内加入24g 30%亚硝酸钠液(含亚硝酸钠0.104mol),控制ph=0.5
‑
2.0、温度t=0
‑
20℃进行重氮化反应1~2小时,用4
‑
二甲氨基苯甲醛的乙醇溶液检测终点(即 5s内不变色)。重氮终点到后,用氨基磺酸消除过量的亚硝酸钠,得到4
‑
乙酰(4'
‑
β
‑
羟乙基砜基硫酸酯苯胺)基苯胺重氮液保存备用。
[0087]
将36.1g(0.1mol)磺化对位酯(2
‑
磺酸
‑4‑
β
‑
羟乙砜硫酸酯苯胺)投入100g水、100g冰中打浆约1h,再加20g 31%盐酸(含hcl 0.17mol),在20
‑
30min内加入24g 30%亚硝酸钠液(含亚硝酸钠0.104mol),控制ph=0.5
‑
2.0、温度t=0
‑
20℃进行重氮化反应1~2小时,用4
‑
二甲氨基苯甲醛的乙醇溶液检测终点(即 5s内不变色)。重氮终点到后,用氨基磺酸消除过量的亚硝酸钠,得到的磺化对位酯重氮液保存备用。
[0088]
(2)偶合:
[0089]
首先将154g(0.1mol)3,5
‑
二羟基苯甲酸加入200g水中进行打浆,控制温度20
‑
25℃,将打浆好的3,5
‑ꢀ
二羟基苯甲酸溶液加入到步骤(1)中得到的4
‑
乙酰(4'
‑
β
‑
羟乙基砜基硫酸酯苯胺)基苯胺中,用30%液碱控制ph在2.0
‑
4.0,温度控制在0
‑
20℃,反应15
‑
20h,用hplc控制游离3,5
‑
二羟基苯甲酸在3%以下即到终点,得下式(b)所示的色基2。
[0090][0091]
将步骤(1)中制得的磺化对位酯重氮液加入到色基2中,用30%液碱控制ph在5.0
‑
8.0,温度t控制在0
‑
20℃,进一步偶合反应5
‑
10h,用h酸试液测试重氮,渗圈处无色即重氮反应完全到终点,得到橙色活性染料化合物(
ⅰ‑
11),水溶液中λmax=420nm。
[0092][0093]
实施例3~63:
[0094]
参照实施例1或实施例2所述的偶氮染料制备方法,采用本领域熟知的中间体原料,经分步重氮和偶合反应,可分别制得下表1中所示结构的染料化合物。
[0095]
表1
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102]
[0103]
[0104]
[0105]
[0106][0107]
应用实施例1
[0108]
取本发明实施例1~63制得的活性染料,分别溶解在水中,加入元明粉50g/l配置成染液。染色浓度 4%(染料对布重),浴比1:20(布重克数对染液体积毫升数),置入棉布60℃下吸附30分钟,加碱(碳酸钠20g/l)固色45分钟。染色织物经水洗、皂煮、干燥,得到黄色至橙色染织物。按iso 105 c10
‑
2006、iso 105x12中的方法分别进行耐洗沾色牢度和摩擦牢度测试,结果如表2:
[0109]
表2
[0110]
[0111][0112]
由表2可知,本发明所述的活性染料化合物耐水洗和耐摩擦牢度优异。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1