用于制备含钼的混合氧化物材料的方法与流程
用于将丙烷氧化为丙烯酸或用于将乙烷氧化脱氢为乙烯的movnbte混合氧化物是现有技术。超过200项专利和大量的科技公开物关注基于movnbte混合氧化物的催化剂。用周期表的其他金属来增强这种混合氧化物是已知的。在此,先前描述过的最高的丙烯酸产率为约60%而乙烯产率为约80%。
基于四种元素的用于催化剂的movnbte基体系首先由三菱提出,用于将丙烷氨氧化成丙烯腈(1989年,ep318295a1)以及氧化成丙烯酸(1994年,ep608838a2)。在jph07-053414(三菱)也说明了通过这种催化剂类型将乙烷氧化氢化为乙烯。
movnbte混合氧化物主要由两种斜方晶系的相组成,被称为“m1”和“m2”(t.ushikubo,k.oshima,a.kayou,m.hatano,studiesinsurfacescienceandcatalysis112,(1997),473)。m1相在选择性氧化反应中展现出重要作用。
根据p.desanto等人,z.kristallogr.219(2004)152,用于选择性氧化的movnbte混合氧化物中的主要相m1和m2例如能够用以下总式描述:
m1:mo1v0.15te0.12nb0.128o3.7或mo7.8v1.2te0.937nb1o28.9
m2:*mo1v0.32te0.42nb0.08o4.6或mo4.31v1.36te1.81nb0.33o19.81
这两种主要相还能以略微不同的化学计量比存在。钒以及钼都在由氧原子形成的八面体的中心并且因此在所述结构中是部分可交换的,使得相同的结构(例如m1相)也可以具有更高的钒含量。在此方面的详细研究可以在p.botella等人,solidstatescience7(2005)507-519中找到。特别地,m2相对于乙烷的氧化脱氢是无活性的(见j.s.valente等人,acscatal.4(2014),1292-1301,第1293页)。因此,对于乙烷的氧化脱氢而言所希望的是由尽可能纯的m1相组成的催化剂。因此尝试以干净且分离的方式制备这些晶相。
ep529853a2公开了一种适合用于从烷烃制备腈的催化剂,其中所述催化剂具有经验式movbtecxxon,其中x为nb、ta、w、ti、al、zr、cr、mn、fe、ru、co、rh、ni、pd、pt、sb、bi、b和ce中的至少一种,b为0.01至1.0,c为0.01至1.0;x为0.01至1.0,且n为如下的数:根据这个数,金属元素的总价数得以满足并且所述催化剂在其x射线衍射图案中在以下的2θ角度下具有x射线衍射峰:衍射角为2θ:22.1°+/-0.3°,28.2°+/-0.3°,36.2°+/-0.3°,45.2°+/-0.3°,50.0°+/-0.3°。
jph07-232071公开了在使用烷烃作为原料以及某种催化剂的情况下在相对较低的温度下且以高产率用于制备腈的方法。所述催化剂的主要成分是由钼、钒、碲、氧和x(x选自铌、钽等的组中的一种或多种元素)形成的混合金属氧化物,其中主要成分(即除了氧以外)的比率通过式i至iv表达:i)0.25<rmo<0.98,ii)0.003<rv<0.50,iii)0.003<rte<0.50,iv)0<rx<0.5,(rmo、rv、rte和rx分别为钼、钒、碲和x的摩尔比例)并且在xrd中,这种混合氧化物的xrd带显示在不同的2θ角度9.0°±0.3°,22.1°±0.3°,27.3°±0.3°,29.2°±0.3°和35.4°±0.3°。据此可以如下地制备腈:在不存在卤化物质的情况在低温下以高产率将烷烃例如与水等在反应体系中进行转化。
用于制备纯m1相的其他成功的实验是基于将m2相从相混合物中溶出。这种实验例如在ep1301457a2、ep1558569a1或wo2009/106474a2中说明。
a.c.sanfiz等人,top.catal.50(2008)19-32描述了水热法合成movnbte氧化物。在这种合成中,仅仅从可溶性化合物出发。通常使用碲酸te(oh)6作为碲的可溶性化合物。在最常见的氧化物型碲化合物teo2中,碲具有+4氧化态。然而二氧化碲(teo2)难溶于水。但在碲酸中,碲具有+6氧化态。即在制备碲酸时,碲必须被氧化成高价。常见的合成通过用过氧化氢氧化氧化碲来进行,这在大规模下带来了安全性问题,因为过氧化氢在自身分解时歧化为水和氧气。因此很难大量制备碲酸。
watanabe(appliedcatal.ageneral,194-195(2000)479-485)尤其说明了从低溶解度的前体moo3、v2o5和teo2进行水热法合成。水热法合成产生了用于氨氧化催化剂的前体,在煅烧之后所述催化剂与通过已知的干式方法制备的催化剂相比具有两倍高的活性。通过固体反应制备的混合氧化物显示出了更低的活性。所建议的是,通过水热合成制备的催化剂的较高活性主要与较大的表面积有关。
在不使用碲酸的情况下,movnbte混合氧化物的合成具有明显成本更低廉的潜在可能性。
在movnbte混合氧化物的合成中使用的nb成分一般是草酸铌铵。相反,氧化铌是难溶的并且因此仅有条件地适合用作初始化合物。
所希望的是一种合成方法,所述合成方法提供干净的movnbte混合氧化物的m1相并且所述合成方法可以用成本低廉的初始化合物(即用简单的金属氧化物,例如三氧化钼、五氧化二钒、五氧化二铌和二氧化碲)来完成。
因此本发明的目的是找到一种简单的、可规模化的、成本低廉且可重现的方法,以在使用二氧化碲且在其他情况下(如果可能)在使用成本低廉的金属氧化物作为初始化合物的情况下以水热法合成来选择性制备movnbte混合氧化物的m1相。
这个目的通过一种用于制备混合氧化物材料的方法实现,所述混合氧化物材料包含元素钼、钒、铌和碲(“movtenb混合氧化物”),所述方法包括以下步骤:
a)制备初始化合物的混合物,所述混合物包含钼、钒、铌和包含碲的初始化合物,在所述包含碲的初始化合物中碲以+4的氧化态存在,
b)在100℃至300℃的温度下以水热法处理所述初始化合物的混合物,以获得产物悬浮液,
c)将从步骤b)产生的产物悬浮液的固体分离出来并干燥,
d)在惰性气体中活化所述固体,以便获得所述混合氧化物材料,
其特征在于,所述包含碲的初始化合物具有小于100μm的粒度d90。
本发明的方法产生了混合氧化物材料,所述混合氧化物材料形成movnbte混合氧化物并且适合用作催化剂材料。
已经发现,在使用例如具有较大颗粒的二氧化碲时,合成只能不完全地进行,即便其他成分都是可溶的金属化合物。产物不能被过滤并且包含很多未转化的纳米颗粒。这形成了可能的钼-钒氧代金属酸盐,其作为具有非常大阴离子的盐而不再溶解,而是形成无法过滤的细小纳米颗粒。
相反,如果为了合成例如使用具有d90<100μm的粒度的二氧化碲,则成功地几乎完全形成了所希望的相。
根据本发明,所使用的包含碲的初始化合物具有d90<100μm、优选d90<75μm、特别优选d90<50μm的粒度。任选地,所使用的二氧化碲可以具有d50<50μm或<35μm的粒度。
此外,包含铌的初始化合物(优选氧化铌)同样可以具有d90<100μm、优选d90<75μm、特别优选d90<50μm的粒度。任选地,所使用的包含铌的初始化合物(优选氧化铌)可以具有d50<50μm或<35μm的粒度。
此外,所使用的所有初始化合物可以具有d90<100μm、优选d90<75μm、特别优选d90<50μm的粒度。任选地,初始化合物可以具有d50<50μm或<35μm的粒度。
初始化合物,例如如此使用的金属氧化物(如二氧化碲),作为粉末存在并且具有粒径分布。粒度d90被定义为在粒径分布中所有颗粒的90%位于其下的颗粒直径限值。中位粒径,即在粒径分布中所有颗粒的一半所低于的粒度,也被称为粒度d50。特别优选地适用的是,用作初始化合物的二氧化碲的粒度d50低于35μm。
所希望的初始化合物的粒度d90或d50可以如下方式获得:从具有较粗颗粒的粒径分布的粉末出发并且将颗粒机械地粉碎。这可以通过研磨来进行,其中可以使用所有适当的且本领域技术人员熟悉的装置,因此例如冲击磨机、行星磨机、研钵等。
所述初始化合物为含有钼、钒、铌和碲的水热合成反应物(前体化合物)。这些反应物分别包含元素钼、钒、铌或碲中的一种或多种。
包含钼的初始化合物可以例如为七钼酸铵或三氧化钼,包含钒的初始化合物可以例如为偏钒酸铵、硫酸氧钒或五氧化二钒,包含铌的初始化合物可以例如为草酸铌铵、草酸铌或氧化铌。根据本发明的包含碲的初始化合物为其中碲以+4氧化态(即作为碲(iv)阳离子)存在的初始化合物,如在二氧化碲中或者式mxn+teo3的化合物(其中n=1或2,并且x=2/n),其中m为碱金属或碱土金属,例如na2teo3。特别优选地,所述包含碲的初始化合物为二氧化碲,它可以任意的水合度存在。
m1相的可能的化学计量比是由文献中充分已知的并且能够通过式mo1vanbbtecox,其中a=0.2至0.3,b=0.1至0.25,c=0.1至0.2且x为取决于金属(mo、v、te和nb)的氧化态而造成电荷平衡的数值。
初始化合物的混合物优选作为含水悬浮液存在并且被水热处理。术语“水热法”涉及用于在水存在下且在提高的温度和/或提高的压力下(例如在高压釜中)制备催化剂材料的反应条件。在此,压力可以在5至30bar、优选10至27bar的范围内。示例性的压力范围为11至15bar,或者约17bar和22至25bar。
通过水热处理(步骤b))获得了包含作为固体的产物的产物悬浮液。在本发明的方法中,在步骤c)中分离出产物悬浮液的固体可以通过一个或多个过滤步骤(例如通过过滤掉母液)来进行。干燥可以在一个步骤中进行或在两个步骤中在流动或静止空气中进行。在此,第一干燥步骤优选在60至150℃(特别优选在80至120℃)下进行,而第二干燥步骤在200至350℃(特别优选在220℃至280℃)下进行。另外,本发明方法的步骤c)可以包含清洗、干燥、煅烧和/或研磨的一个或多个步骤。煅烧可以在200至500℃下、优选250℃至350℃下在空气中进行。
在步骤c)中的滤液的干燥之后,将干燥的混合物例如在流动的或静止的惰性气体氛围中在大约500至700℃下活化至少1小时(步骤d)。尤其氮气、氦气或氩气适合用作惰性气体。优选的是,所述活化在550℃至650℃范围内进行。例如,活化可以在大约600℃下进行大约2小时。
所获得的movnbte混合氧化物可以用作用于将烃类氧化和/或氧化脱氢的催化剂材料,尤其用于将丙烷选择性氧化为丙烯酸或者将乙烷氧化脱氢为乙烯。所述催化剂材料典型具有5至25m2/g的bet表面积。
根据本发明方法制备的所获得的催化剂材料能以不同方式用于商业催化剂中。例如可以通过压片而被加工成催化剂片料,然后可以将其填充到反应器中。
所述催化剂材料还可以与适合的粘结剂一起被加工成挤出物(片料、成型体、蜂窝体等)。可以使用本领域技术人员熟悉的且表现为适合的粘结剂作为粘结剂。优选的粘结剂尤其为伪勃姆石以及硅酸盐类粘结剂,如胶体二氧化硅或二氧化硅溶胶。
所述催化剂材料另外可以与其他成分一起、优选与粘结剂、特别优选与有机粘结剂(例如有机粘合剂、聚合物、树脂或蜡)一起被加工成载体涂层(washcoat),所述载体涂层可以施加到金属或陶瓷载体上。在适当情况下可以进行额外的浸渍步骤或煅烧步骤。
所获得的movnbte混合氧化物通过以下分析方法来表征:
根据本发明方法形成的本发明movnbte混合氧化物的x射线衍射图具有如下的衍射反射(beugungsreflex)h、i、k和l,其顶点位于衍射角度(2θ)26.2°±0.5°(h),27.0°±0.5°(i),7.8°±0.5°(k)和28.0°±0.5°(l),其中衍射反射h、i、k和l的强度ph、pi、pk、pl可以满足以下关系,其中rx(x=1至3)为由这些关系定义的强度比:
r1=ph/(ph+pi)>0.3,优选>0.35且特别优选>0.4;和/或
r2=pi/(pi+pl)>0.5,优选>0.6且特别优选>0.63;和/或
r3=pi/(pi+pk)<0.8,优选<0.75且特别优选<0.7。
在所获得的movnbte混合氧化物的实施方式的x射线衍射图中,衍射反射i可以具有第二大的强度和/或衍射反射h可以具有第三大的强度。
所获得的movnbte混合氧化物在实施例中作为催化剂材料使用并且因此在实验信息中部分地标识为催化剂。
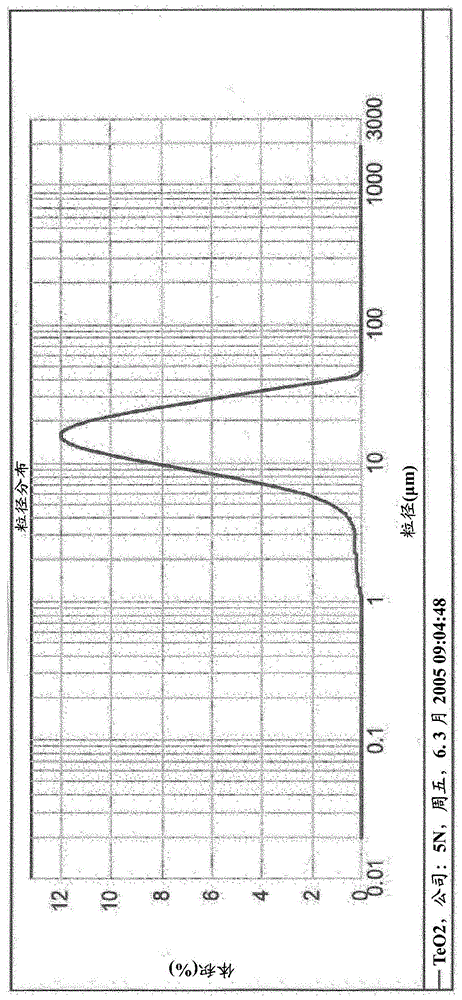
图1:实施例1中所使用的teo2的粒径分布,其中粒度值d10=7.625μm,d50=15.140μm,d90=27.409μm。
图2:来自实施例1的movnbte混合氧化物的xrd。
图3:对比实施例1中所使用的teo2的粒径分布,其中粒度值d10=16.45μm,d50=43.46μm,d90=236.48μm。
图4:来自对比实施例1的movnbte混合氧化物的xrd。
图5:实施例2中所使用的teo2的粒径分布。
图6:来自实施例2的混合氧化物材料的xrd。
图7:实施例3中所使用的nb2o5在研磨之前和之后的粒径分布对比。
图8:来自实施例3的movnbte混合氧化物的xrd。
图9:来自对比实施例3的movnbte混合氧化物的xrd。
表征方法:
为了测定所获得的movnbte混合氧化物的参数,使用了以下方法:
1.bet表面积
测定根据din66131按照bet方法进行;bet方法的公开内容也可以在j.am.chem.soc.60,309(1938)中找到。将待测定的样品在u形石英反应器中在200℃下在ar气氛中干燥(f=50ml(分钟),持续1.5小时)。然后将反应器冷却到室温、抽真空并且在杜瓦瓶中用液氮浸没。在77k下用rxm100吸附系统(advancedscientificdesign,inc.)执行氮气吸附。
对于相应的movnbte混合氧化物样品,在200℃下真空中干燥的材料上进行bet表面积测定。在当前关于movnbte混合氧化物的bet表面的说明中的信息也是关于分别使用的催化剂材料的bet表面积(在200℃下在真空中干燥)。
2.粉末x射线衍射测量法(xrd)
通过粉末x射线衍射测量法(xrd)来建立x射线衍射图并且根据scherrer公式来进行分析。对在600℃下在氮气中活化的催化剂材料测量xrd谱。
在philips基于pw3710型的pw1050布拉格-布伦塔诺仲聚焦测角仪中,在40kv和35ma下在使用cu-kα辐射(波长=0.15418nm)、石墨单色器和比例计数器的情况下进行测量。xrd扫描以0.04°的步幅(2塞塔,2θ)以数字方式记录。为了对相进行定量分析,加入sic作为内标。在此加入约5%的sic,但是其量被准确称量。这个量在相分析中给出。根据rietveld方法用topas软件进行相分析。这种相分析的结果在xrd图中给出。所希望的m1相的准确量可以如下方式计算:m1相对整个样品的比例(如所给出的)是相对于没有sic的样品。
3.粒度
用激光散射方法测定粒径分布。为此使用malvernmastersizer2000。分析根据fraunhofer方法进行。
现应借助于下文中的不应理解为限制性的实施例来详细解说本发明。
实施例:
实施例1
在高压釜(40l)中放入3.3l蒸馏水并且在搅拌下加热到80℃。在此期间添加725.58g的四水合七钼酸铵(来自hcstarck)并且溶解(ahm溶液)。在两个5l玻璃烧杯中分别将1.65l蒸馏水在磁力搅拌器的搅拌下通过控温同样加热到80℃。然后在这些玻璃烧杯中分别加入405.10g的水合硫酸氧钒(来自gfe,v含量:21.2%)、185.59g草酸铌铵(hcstarck,nb含量:20.6%)并且溶解(v溶液和nb溶液)。
相继将v溶液泵送到ahm溶液中,然后加入65.59g作为固体的teo2粉末(5n+的teo2,粒径分布见图1)和1.65l蒸馏水,在80℃下继续搅拌1小时并且最终借助于软管泵将nb溶液泵送到ahm溶液中。泵送时间:v溶液:4.5分钟,190rpm(软管直径:8x5mm),nb溶液:6分钟,130rpm(软管直径:8x5mm)。
所产生的悬浮液在80℃下继续搅拌10分钟。在沉淀时搅拌器的速度为90rpm。
随后用氮气覆盖,其方式为,在带有氮气的高压釜中升高压力直至约6bar并且将泄放阀打开,直至在压力下n2流过高压釜(5分钟)。在结束时通过排气阀将压力再次泄放到直至1bar的残余压力。
在40l高压釜中在175℃下通过锚式搅拌器在90rpm的搅拌速度下进行水热法合成20小时(升温时间:3小时)。
在合成之后借助于真空泵通过蓝带过滤器过滤并且将滤饼用5l蒸馏水清洗。
在80℃下在干燥箱中干燥3天并且随后在冲击磨机中研磨,其中获得了0.8kg的固体产量。
在空气流中在280℃下煅烧4小时(加热速率5℃/分钟,空气:1l/分钟)。
在蒸馏器中在600℃下在n2通流中活化2小时(加热速率5℃/分钟,n2:0.5l/分钟)。
所使用的teo2的粒径分布为:
d10=7.625μmd50=15.14μmd90=27.409μm
产物的分析表征:
bet=15m2/g
xrd:
来自实施例1的混合氧化物材料的xrd在图2中示出并且具有以下相分布:
m1=90.50%,
m2=2.82%
(mo0.9v1.1)o5=1.15%
sic(标准)=5.53%
对比实施例1:
在高压釜(40l)中放入6.6l蒸馏水并且在搅拌下加热到80℃。在此期间添加1451.16g的四水合七钼酸铵(hcstarck)并且溶解(ahm溶液)。在两个5l玻璃烧杯中分别将3.3l蒸馏水在磁力搅拌器的搅拌下通过控温同样加热到80℃。然后在这些玻璃烧杯中分别加入810.21g的水合硫酸氧钒(gfe,v含量:21.2%)、370.59g草酸铌铵(hcstarck,nb含量:20.6%)并且溶解(v溶液和nb溶液)。
相继将v溶液泵送到ahm溶液中,然后加入131.18g作为固体的teo2粉末(alfaaesar,粒径分布见图3)和3.3l蒸馏水,在80℃下继续搅拌1小时并且最终借助于软管泵将nb溶液泵送到ahm溶液中。泵送时间:v溶液:5分钟,290rpm(软管直径:8x5mm),nb溶液:5分钟,275rpm(软管直径:8x5mm)。
所产生的悬浮液在80℃下继续搅拌10分钟,在沉淀时搅拌器的速度为90rpm。
随后用氮气覆盖,其方式为,在带有氮气的高压釜中升高压力直至约6bar并且将泄放阀打开,直至在压力下n2流过高压釜(5分钟)。在结束时通过排气阀将压力再次泄放到直至1bar的残余压力。
在40l高压釜中在175℃下通过锚式搅拌器在90rpm的搅拌速度下进行水热法合成20小时(升温时间:3小时)。
在合成之后借助于真空泵通过蓝带过滤器过滤并且将滤饼用5l蒸馏水清洗。过滤持续多日。
在80℃下在干燥箱中干燥3天并且随后在冲击磨机中研磨,其中获得了0.5kg的固体产量。
对比实施例1的产量仅为本发明实施例的一半左右。在空气流中在280℃下煅烧4小时(加热速率5℃/分钟,空气:1l/分钟)。
在蒸馏器中在600℃下在n2通流中活化2小时(加热速率5℃/分钟,n2:0.5l/分钟)。
所使用的teo2的粒度值:
d10=16.45μmd50=43.46μmd90=236.48μm
来自对比实施例1的movnbte混合氧化物的xrd在图4中示出并且具有以下相分布:
m1=51.88%
m2=8.12%
(mo0.9v1.1)o5=12.51%
(v0.35mo4.65)o14=23.19%
sic(标准)=3.59%
实施例2
在高压釜(40l)中放入3.3l蒸馏水并且在搅拌下加热到80℃。在此期间添加725.58g的四水合七钼酸铵(hcstarck)并且溶解(ahm溶液)。在两个5l玻璃烧杯中分别将1.65l蒸馏水在磁力搅拌器的搅拌下通过控温同样加热到80℃。然后在这些玻璃烧杯中分别加入405.10g的水合硫酸氧钒(gfe,v含量:21.2%)、185.59g草酸铌铵(hcstarck,nb含量:20.6%)并且溶解(v溶液和nb溶液)。
在前一天将65.59g的teo2(alphaaesar,来自对比实施例1)在200g蒸馏水中研磨3小时(retsch的球磨pm100)并且用1.45l蒸馏水转移到玻璃烧杯中(研磨之后的粒度见图5)。
相继将v溶液泵送到ahm溶液中,然后加入前一天研磨的te悬浮液,在80℃下继续搅拌1小时并且最终借助于软管泵将nb溶液泵送到ahm溶液中。泵送时间:v溶液:5分钟,290rpm(软管直径:8x5mm),nb溶液:5分钟,275rpm(软管直径:8x5mm)。
所产生的悬浮液在80℃下继续搅拌10分钟,在沉淀时搅拌器的速度为90rpm。
随后用氮气覆盖,其方式为,在带有氮气的高压釜中升高压力直至约6bar并且将泄放阀打开,直至在压力下n2流过高压釜(5分钟)。在结束时通过排气阀将压力再次泄放到直至1bar的残余压力。
在40l高压釜中在175℃下通过锚式搅拌器在90rpm的搅拌速度下进行水热法合成20小时(升温时间:3小时)。
在合成之后借助于真空泵通过蓝带过滤器过滤并且将滤饼用5l蒸馏水清洗。
在80℃下在干燥箱中干燥3天并且随后在冲击磨机中研磨,其中实现了0.8kg的固体产量。
在空气流中在280℃下煅烧4小时(加热速率5℃/分钟,空气:1l/分钟)。
在蒸馏器中在600℃下在n2流中活化2小时(加热速率5℃/分钟,n2:0.5l/分钟)。
研磨3小时的teo2的粒度值:
d10=0.569μmd50=2.992μmd90=6.326μm
产物的分析表征:
bet=12m2/g
来自实施例2的movnbte混合氧化物的xrd在图6中示出并且具有以下相分布:
m1=86.30%
m2=2.78%
(mo0.9v1.1)o5=0.75%
(v0.35mo4.65)o14=3.75%
sic(标准)=5.01%
实施例3:
首先将teo2(alfaaesar,来自对比实施例1)悬浮在200g蒸馏水中并且在球磨(如在实施例2中一样)中研磨。随后将这部分用500ml蒸馏水转移到玻璃烧杯中。将nb2o5悬浮在200g蒸馏水中并且在相同的球磨中研磨。在图7中示出了在研磨之前和之后的粒径分布对比。
随后将这部分用500ml蒸馏水转移到玻璃烧杯中。第二天早晨将其加热到80℃,向nb2o5悬浮液中加入107.8g的二水合草酸并且搅拌约1小时。在高压釜(40l)中放入6l蒸馏水并且在搅拌下加热到80℃。在水已经达到这个温度之后,向草酸中相继加入61.58g的柠檬酸、19.9g的乙二醇、615.5g的moo3(sigmaaldrich),124.5g的v2o5,经研磨的teo2和经研磨的nb2o5。使用850ml蒸馏水来转移和清洗容器。在高压釜中全部的水量为8.25l,搅拌器的速度为90rpm。然后用氮气覆盖。在40l的高压釜中在190℃下进行水热法合成48小时。在合成之后借助于真空泵通过蓝带过滤器过滤并且将滤饼用5l蒸馏水清洗。
在80℃下在干燥箱中干燥3天并且随后在冲击磨机中研磨,其中实现了0.8kg的固体产量。
在空气流中在280℃下煅烧4小时(加热速率5℃/分钟,空气:1l/分钟)。
在蒸馏器中在600℃下在n2流中活化2小时(加热速率5℃/分钟,n2:0.5l/分钟)。
来自实施例3的movnbte混合氧化物的xrd在图8中示出并且具有以下相分布:
m1=85.79%
m2=1.95%
(mo0.9v1.1)o5=1.43%
moo3=3.31%
nb2o5=2.86%
sic(标准)=4.66%
对比实施例2:
首先将teo2(alfaaesar,来自对比实施例1)悬浮在200g蒸馏水中并且在球磨(如在实施例2中一样)中研磨,然后用水转移到玻璃烧杯中,使得玻璃烧杯中的水体积为1650ml。
在高压釜(40l)中放入6.6l蒸馏水并且在搅拌下加热到80℃。一旦水已经到达这个温度,就相继加入61.58g的柠檬酸、194g的二水合草酸、19.9g的乙二醇、615.5g的moo3(sigmaaldrich),124.5g的v2o5,经研磨的teo2和56.8g的nb2o5(未研磨情况下具有来自图7的粒径分布,也具有超过100μm的颗粒)。然后用氮气覆盖。在40l的高压釜中在190℃下进行水热法合成48小时。在合成之后借助于真空泵通过蓝带过滤器过滤并且将滤饼用5l蒸馏水清洗。
在80℃下在干燥箱中干燥3天并且随后在冲击磨机中研磨,其中实现了0.8kg的固体产量。
在空气流中在280℃下煅烧4小时(加热速率5℃/分钟,空气:1l/分钟)。在蒸馏器中在600℃下在n2流中活化2小时(加热速率5℃/分钟,n2:0.5l/分钟)。
来自对比实施例3的movnbte混合氧化物的xrd在图9中示出并且具有以下相分布:
m1=17.34%
m2=1.75%
(v0,35mo4,65)o14=34.35%
movo5=24.57%
temo6o16=17.39%
sic(标准)=4.6%
可以清楚地看到,使用先前未能与草酸反应的未研磨的氧化铌,仅实现了17%的m1相。
- 还没有人留言评论。精彩留言会获得点赞!