一种铟基金属有机框架/类石墨相碳化氮纳米片复合材料及其制备方法与应用与流程
本发明涉及一种铟基金属有机框架/类石墨相碳化氮纳米片复合材料及其制备方法与应用,具体属于光催化材料制备领域。
背景技术:
药物是维护公共健康和生活质量所必不可少的,近年来,随着社会的发展及生活水平的提高,药物的使用量逐年上升,随之而来的是大量残留的药物经制药厂、医疗垃圾、废水及排泄物等途径进入环境中,对生态环境及人体健康形成了潜在的威胁。布洛芬是全球环境中检出频率最高的非甾体抗炎药之一,由于其毒性低,疗效和副作用均优于阿司匹林和扑热息痛而得以在世界范围内广泛应用。布洛芬在生产和使用过程中会通过多种途径进入环境,成为一种环境中土壤、地表水、地下水、海水普遍存在的典型类污染物。由于其自身独特的化学结构和特性,布洛芬在环境中能稳定存在,污水厂的传统工艺不能有效去除,从而导致其在环境及生物体内积累,有研究表明,长期摄入痕量水平的布洛芬会引起生物畸形和微生物的抗药性等。因此,制备新型可见光催化材料,实现对布洛芬的高效彻底降解,对解决饮用水安全问题及维护良好的水生态环境具有重大的现实意义。
金属有机骨架材料(metal-organicframeworks,mofs)是近年来研究十分热门的一种新型纳米网状材料。因为其具有超高的比表面积、高空隙率、孔径和功能可调等优点,已经涉及不同领域的应用。而其中具有光催化活性中心的mof兼具mof的优点与良好的可见光催化性能,展现出卓越的降解有机污染物的能力。然而,单纯的mof在光催化反应的过程中,光生电子和空穴非常容易发生复合,从而导致其光催化活性的降低。因此,进一步提高mof材料的光催化活性是研究的重点。
通过将mof与其他能带匹配的材料进行复合,在材料之间构建异质结结构是拓宽mof可见光响应范围和减小光生电子和空穴重组几率的有效方法。通过调研发现,类石墨相氮化碳是满足上述条件的理想材料。作为一种新型非金属光催化材料,类石墨相碳化氮具有较窄的带隙(约为2.7ev),能吸收波长小于475nm的可见光,在普通可见光下就能起到光催化作用。同时,类石墨相碳化氮还具有易制备,化学稳定性及热稳定性好,对环境友好,无二次污染,成本低廉等优势,近年来,被广泛应用于室内空气污染治理和有机物降解。目前,已经有部分相关研究,如:g-c3n4/mil-53(al)(appliedorganometallicchemistry,2015,29(10):690–697),g-c3n4/mil-125(ti)(appliedcatalysisb:environmental,2015,174-175(3):445-454)。但都存在着g-c3n4易团聚,在mof中分散不均匀的问题,影响了材料间光生电子和空穴的传递,从而限制了材料光催化活性的提升。因此,本发明围绕解决上述问题展开。
技术实现要素:
本发明的首要目的在于提供一种铟基金属有机框架/类石墨相碳化氮纳米片复合材料。
本发明另一个目的在于提供上述铟基金属有机框架/类石墨相碳化氮纳米片复合材料的制备方法。
本发明的再一目的在于提供上述铟基金属有机框架/类石墨相碳化氮纳米片复合材料在光催化领域的应用。
本发明的目的通过以下技术方案实现:
一种铟基金属有机框架/类石墨相碳化氮纳米片复合材料的制备方法,包括以下步骤:
(1)制备类石墨相碳化氮纳米片;
(2)采用下述任意项方法制备铟基金属有机框架/类石墨相碳化氮纳米片复合材料:
a.将类石墨相碳化氮纳米片分散于n,n′-二甲基甲酰胺中,搅拌形成均匀的淡黄色悬浊液后,超声处理1-10h使其进一步分散;再向上述分散液中同时加入硝酸铟水合物和2-氨基对苯二甲酸并重复上述搅拌和超声操作;
b.将类石墨相碳化氮纳米片和硝酸铟水合物分散于n,n′-二甲基甲酰胺中,搅拌形成均匀的淡黄色悬浊液后,超声处理1-10h使其进一步分散;再向上述分散液中加入2-氨基对苯二甲酸并重复上述搅拌和超声操作;
c.将类石墨相碳化氮纳米片、硝酸铟水合物和2-氨基对苯二甲酸加入n,n′-二甲基甲酰胺中,搅拌形成均匀的淡黄色悬浊液后,超声处理4-16h使其进一步分散;
(3)将步骤(2)所得的混合液于110-140℃下连续加热6h-24h,冷却后抽滤得初产物;
(4)将步骤(3)所得初产物用溶剂洗涤纯化,真空干燥后,得最终产物。
优选地,步骤(2)类石墨相碳化氮纳米片在混合液中的含量为0.004g/ml-0.008g/ml,硝酸铟在混合液中的含量为0.08-0.12g/ml,其中硝酸铟与2-氨基对苯二甲酸的质量比为(4.8-5.3):1。
优选地,步骤(2)a和b中超声处理时间为2-8h,搅拌时间为1-4h;c中超声处理时间为8-12h,搅拌时间为3-6h。
优选地,步骤(4)所述溶剂为n,n′-二甲基甲酰胺和甲醇;所述的纯化为分别用n,n′-二甲基甲酰胺和甲醇洗涤3次。
优选地,步骤(4)所述真空干燥的温度为100-130℃,时间为12-24h。
优选地,所述类石墨相碳化氮纳米片的制备:将三聚氰胺粉末用锡纸密封后置于通n2的管式炉中于450-550℃下煅烧4-6h得淡黄色块状产物,研磨成粉末后再转移至马弗炉中于500-600℃下高温热氧化2-6h,得类石墨相碳化氮纳米片。
优选地,所述管式炉升温速率为5℃/min。
上述方法制备得到的铟基金属有机框架/类石墨相碳化氮纳米片复合材料在光催化降解水体中残留药物的应用。
上述铟基金属有机框架/类石墨相碳化氮纳米片复合材料在降解水体中布洛芬中的应用。
与现有技术相比,本发明具有以下优点:
(1)本发明将热聚合法制备的类石墨相碳化氮通过再次高温热氧化,得到类石墨相碳化氮纳米片,在提高材料比表面积和光催化性能的同时,有效改善了类石墨相碳化氮易团聚的问题。
(2)通过溶剂热法原位引入类石墨相碳化氮纳米片,使其与铟基金属有机框架晶体界面紧密结合且均匀分布,形成异质结,有利于材料间光生电子和空穴的快速传递,降低其重组几率,拓宽可见光吸收范围,显著提高材料的光催化活性。
(3)最终制备的铟基金属有机框架/类石墨相碳化氮纳米片复合材料成本低廉,工序简单,有望实现大规模生产。且复合材料在保持mof优异的吸附能力的同时,能作为一种有效的光催化剂,在彻底降解含布洛芬废水方面效果显著。
附图说明
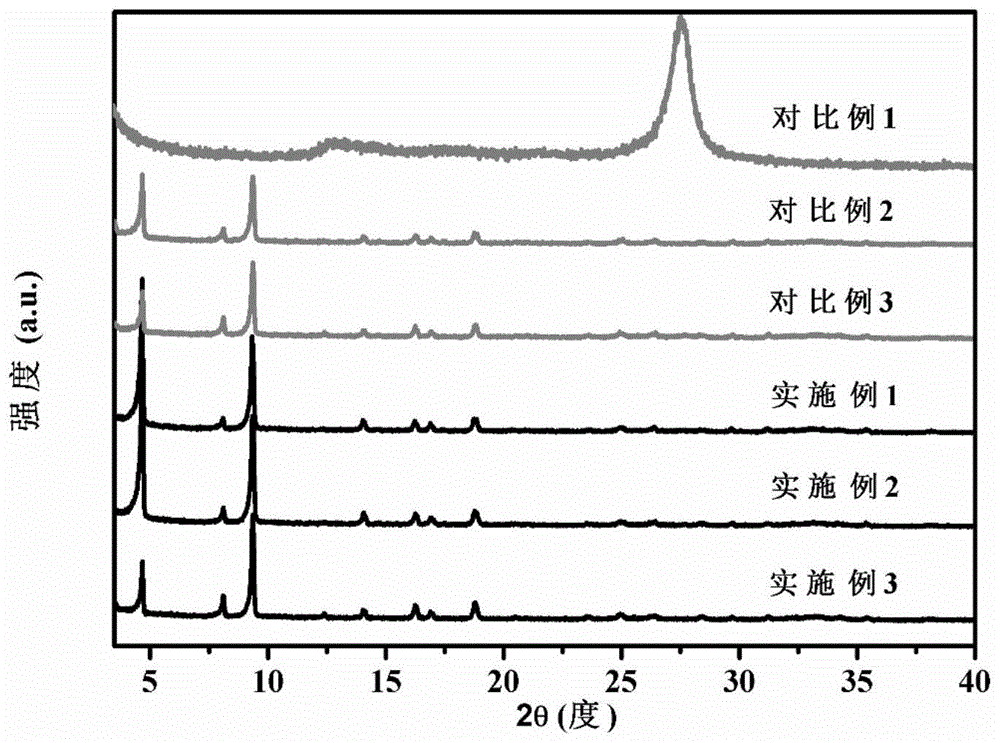
图1是对比例1所制得的g-c3n4纳米片、对比例2所制得的mil-68(in)-nh2及对比例3所制得的g-c3n4/mil-68(in)-nh2复合材料与实施例1~3所制得的g-c3n4/mil-68(in)-nh2复合材料的xrd谱图对比。
图2是对比例1所制得的g-c3n4纳米片、对比例2所制得的mil-68(in)-nh2及对比例3所制得的g-c3n4/mil-68(in)-nh2复合材料与实施例1~3所制得的g-c3n4/mil-68(in)-nh2复合材料的ftir谱图对比。
图3是不同光催化剂在可见光下(λ>420nm)对布洛芬的降解曲线。(实验条件:光催化剂用量为20mg、布洛芬用量为200ml、布洛芬初始浓度为20mg/l)。
具体实施方式
下面结合实施例和附图对本发明作进一步描述,但本发明的实施不限于此,任何变化实施都包含在本发明的技术范围内。
对比例1
g-c3n4纳米片的制备
准确称取10g三聚氰胺粉末于瓷舟中,用锡纸密封后置于通n2的管式炉中于550℃下煅烧4h(升温速率为5℃/min)得淡黄色块状产物,研磨成粉末后转移至马弗炉中于500℃下再加热2h,即得类石墨相碳化氮纳米片。
对比例2
mil-68(in)-nh2的制备
将0.578gin(no3)3.xh2o和0.118g2-氨基对苯二甲酸同时加入6.2mln,n′-二甲基甲酰胺中,剧烈搅拌30min后,超声10min得到均匀溶液。将上述溶液转移至50ml高压反应釜中,于125℃下加热24h,自然冷却后得初产物。用50mln,n′-二甲基甲酰胺和甲醇分别洗涤三次,使材料进一步纯化。最后,置于真空干燥箱内于130℃下干燥活化12h,用玛瑙研钵磨细即得mil-68(in)-nh2淡黄色粉末。
对比例3
g-c3n4/mil-68(in)-nh2的制备
准确称取0.02g类石墨相碳化氮纳米片分散于5mln,n′-二甲基甲酰胺中,剧烈搅拌1h形成均匀的淡黄色悬浊液后,超声处理2h使其进一步分散均匀。向溶液中同时加入0.578g硝酸铟水合物、0.118g2-氨基对苯二甲酸以及1.5mln,n′-二甲基甲酰胺,并重复上述搅拌和超声操作。将所得的混合液转移至50ml高压反应釜中,于125℃下连续加热24h,冷却至室温后抽滤得初产物。初产物用50mln,n′-二甲基甲酰胺和甲醇分别洗涤三次,置于真空干燥箱内干燥12h,即得铟基金属有机框架/类石墨相碳化氮纳米片复合材料。
实施例1
g-c3n4/mil-68(in)-nh2的制备
准确称取0.04g类石墨相碳化氮纳米片分散于6.5mln,n′-二甲基甲酰胺中,剧烈搅拌1.5h形成均匀的淡黄色悬浊液后,超声处理3h使其进一步分散均匀。向溶液中同时加入0.578g硝酸铟水合物和0.118g2-氨基对苯二甲酸,并重复上述搅拌和超声操作。将所得的混合液转移至50ml高压反应釜中,于130℃下连续加热18h,冷却至室温后抽滤得初产物。初产物用50mln,n′-二甲基甲酰胺和甲醇分别洗涤三次,置于真空干燥箱内干燥24h,即得铟基金属有机框架/类石墨相碳化氮纳米片复合材料。
实施例2
g-c3n4/mil-68(in)-nh2的制备
准确称取0.08g类石墨相碳化氮纳米片和1.245g硝酸铟水合物分散于13mln,n′-二甲基甲酰胺中,剧烈搅拌2h形成均匀的淡黄色悬浊液后,超声处理4h使其进一步分散均匀。再向溶液中加入0.236g2-氨基对苯二甲酸,并重复上述搅拌和超声操作。将所得的混合液转移至50ml高压反应釜中,于110℃下连续加热12h,冷却至室温后抽滤得初产物。初产物用100mln,n′-二甲基甲酰胺和甲醇分别洗涤三次,置于真空干燥箱内干燥24h,即得铟基金属有机框架/类石墨相碳化氮纳米片复合材料。
实施例3
g-c3n4/mil-68(in)-nh2的制备
准确称取0.12g类石墨相碳化氮纳米片、1.743g硝酸铟水合物和0.345g2-氨基对苯二甲酸分散于15mln,n′-二甲基甲酰胺中,剧烈搅拌4h形成均匀的淡黄色悬浊液后,超声处理6h使其彻底分散均匀。将所得的混合液转移至50ml高压反应釜中,于140℃下连续加热6h,冷却至室温后抽滤得初产物。初产物用100mln,n′-二甲基甲酰胺和甲醇分别洗涤三次,置于真空干燥箱内干燥24h,即得铟基金属有机框架/类石墨相碳化氮纳米片复合材料。
本发明的对比例1~3和实施例1~3所制得的g-c3n4纳米片、mil-68(in)-nh2和g-c3n4/mil-68(in)-nh2,其xrd和光催化降解布洛芬曲线如下:
(1)xrd图谱分析
对本发明的对比例1~3以及实施例1~3所制得材料的晶体结构采用荷兰帕纳科公司生产的empyrean锐影x射线衍射仪进行具体分析,其中操作条件为:铜靶,40kv,40ma,步长0.0131度,扫描速度9.664秒/步,结果如图1所示。
根据图1分析可得,实施例1~3所制得的g-c3n4/mil-68(in)-nh2复合材料的衍射峰与对比例2所制得的mil-68(in)-nh2及对比例3所制得的g-c3n4/mil-68(in)-nh2复合材料的xrd衍射图谱基本一致,且峰型尖锐,无新杂峰出现,这表明实施例1~3所制得的g-c3n4/mil-68(in)-nh2复合材料保留了母体mil-68(in)-nh2的晶体结构且晶型良好。
(2)ftir图谱分析
采用傅里叶转换红外光谱表征手段对所制备材料的表面基团进行分析,操作步骤为:按质量比为100:1称取一定量kbr粉末和样品,研磨至混合均匀,置于压片机下制成圆形薄片,固定于仪器中进行光谱扫描,扫描波长范围为:500-4000cm-1。根据图2结果,对比例1中波数在1650-1200cm-1范围内的振动带对应于g-c3n4典型的芳香族c-n伸缩振动和七嗪环的平面外弯曲振动,而881和810cm-1处的吸收峰则与三嗪环单元的呼吸模式有关;对比例2中位于1255cm-1处的吸收峰是c-n的伸缩振动模式;对比例3与实施例1-3的红外谱图中出现了对比例1-2所有的特征振动峰,证明g-c3n4与mil-68(in)-nh2的成功复合,且复合材料中保留了单体材料完整的有机官能团。
(3)对布洛芬的降解效果分析
通过布洛芬的降解实验验证对比例1~3及实施例1~3材料的光催化性能,实验条件如下:
光催化剂投加量为0.1g/l;布洛芬初始浓度为20mg/l;
吸附时间为1h;光照时间为2h;光源为300w氙灯(λ>420nm);
检测仪器为waters高效液相色谱(hplc)4.6mm×250mm5μmc18柱;
检测条件为流动相0.1%甲酸水溶液/乙腈=30:70(v/v),检测波长223nm。
色谱柱温度为40℃,进样量为10μl,出峰时间5.5min。
图3是实施例1~3制得的g-c3n4/mil-68(in)-nh2复合材料与对比例1制得的g-c3n4纳米片、对比例2制得的mil-68(in)-nh2及对比例3制得的g-c3n4/mil-68(in)-nh2复合材料对布洛芬的光催化降解曲线图。由图3可得:与对比例1~3相比,实施例1~3所制得的g-c3n4/mil-68(in)-nh2复合材料对水体中布洛芬的光催化降解效果更为显著,经过120min的光照后,实施例2对布洛芬的光催化去除效率高达97.2%,分别为对比例1和对比例2的10.8倍和1.6倍。因此,可以初步证明,本发明所制得的g-c3n4/mil-68(in)-nh2复合材料对可见光的利用效率更高,光生电子和空穴能实现快速分离,与单体材料相比,复合材料的光催化活性得到极大提升。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所做的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!