烷烃脱氢制烯烃催化剂及其制备和脱氢方法
本发明属于化学合成领域,具体涉及一种烷烃脱氢制备烯烃的催化剂及脱氢方法。
背景技术:
烯烃是重要的化工原料,例如苯乙烯作为芳烃的衍生物,是化工行业生产高聚物重要的单体原料,被广泛应用于和人们日常生活密切相关的领域中。据估计,每年苯乙烯相关的工业产值可达600亿美元。近20年来,随着全球苯乙烯下游产品市场的不断开拓,苯乙烯需求量逐年上升。虽然从国际市场来说苯乙烯的产能有所富裕,但我国作为苯乙烯需求增速最快的国家之一,苯乙烯自给率仍处于较低水平。相关数据显示,到2022年我国苯乙烯生产能力将超过1500万吨/年,而根据目前己知的下游装置新、扩、拟建计划,下游装置苯乙烯表观需求量将达到1800~1900万吨/年,缺口仍然超过300万吨/年。在反倾销、贸易战的大背景下,我国苯乙烯进口附加税大幅上涨,这意味着开发新的苯乙烯生产技术,尤其是自主研发高性能、低成本、低能耗的乙苯脱氢制苯乙烯催化剂,对于提高国内苯乙烯自给率具有重要的意义又富有挑战性。
在实际生产过程中,考虑到乙苯脱氢制备苯乙烯的反应为分子数增加的强吸热反应,受热力学平衡限制较大,低压和高温条件下有利于提高反应转化率。1937年,美国陶氏化学公司(dow)和德国巴斯夫公司(basf)同时实现了乙苯脱氢制取苯乙烯的工业化生产。经过80多年的发展,目前工业上应用的苯乙烯生产方法主要有两种:乙苯直接脱氢法和苯乙烯环氧乙烷联产法,而乙苯催化脱氢催化剂也已由初期使用的锌系、镁系催化剂逐步被综合性能更为优异的铁系催化剂所取代。当前,使用含钾氧化铁或氧化锌作为催化剂、在600~650℃的高温和过量水蒸气(水/烃比7~15)保护下催化乙苯直接脱氢生产苯乙烯的lummus/monsanto/uop工艺是现目前世界上应用最为广泛的苯乙烯生产技术,该项技术研究至今已有70多年的历史,相应的研究成果也得到了广泛的认可,采用该工艺生产的苯乙烯量占全球苯乙烯生产总量的90%以上。但水蒸气的大量消耗,使得整个生产过程能耗巨大。此外,考虑到温度过高会加剧苯乙烯的聚合,且钾在高温下不稳定,容易向低温区或催化剂颗粒中心转移,导致催化剂的稳定性变差。
目前,国外最大的乙苯催化脱氢催化剂供应商是德国南方化学公司和美国标准催化剂公司,德国巴斯夫公司和陶氏化学公司也仍在生产该类催化剂。就南方化学公司来说,其新推出的styromax系列催化剂已取代了早期生产的g-64、g-84和g-97系列催化剂。美国标准催化剂公司的乙苯催化脱氢催化剂也开发较早,尤其是shell-105曾长期应用于各大型生产装置。近年来,标准公司为适应生产装置的要求,又相继推出c-035、c-045、c-145和flexicatyellow/blue等催化剂。我国乙苯催化脱氢催化剂的自主研究始于20世纪60年代,中田石油集团石油化工研究院兰州化工研究中心研发的lh系列和厦门大学的xh系列、中科院大连化学物理研究物所开发的dc系列和中国石化上海石油化工研究院开发的gs系列催化剂均已实现工业应用。如:cn106423187a、cn106423240a、cn105312059a及cn105749934a。特别是专利cn105749934a报道的催化剂可在相对较低的水/烃(1.2~2.0)比下使用,但引入水蒸气时的高能耗问题仍然存在。此外,以上体系的催化剂,均以铈、铬、锰等金属的氧化或稀土元素做助剂,它们中的部分物质,如铬氧化物既提高了催化剂成本,也对环境甚至人类健康都有较大危害。另一方面,催化脱氢工艺的改进,如提高单程转化率和降低热能的使用己接近极限,而氧化脱氢体系中则存在苯乙烯深度氧化、反应选择性不高以及实际操作中反应物组成受爆炸极限范围的限制等问题,也从一定程度上限制了脱氢反应的转化率。在上述背景下,开发高转化率、高选择性、高稳定性和环境友好型的催化剂,尤其是开发出适用于无水或低水/烃比条件下操作的催化剂,对苯乙烯生产过程中的节能降耗具有重要意义。
近年来,非金属催化剂的探索成为研究的热点。特别是具有规整结构的、元素多变的二维材料的出现,为烷烃脱氢催化剂的研究,提供了更多的可能性。2010年,中国科学院沈阳金属研究所苏党生课题组首次报道了纳米金刚石(nd)可作为无水蒸气条件下催化乙苯直接脱氢制备苯乙烯的高效非金属催化剂,并认为具有较高氧原子电子密度的不饱和酮/二酮型羰基(c=o)是该催化剂的活性中心。这项工作掀起了有关碳基催化剂应用于乙苯脱氢反应的研究热潮。之后,中科院沈阳金属所以及大连理工大学在此基础上,又陆续报道了碳纳米管(新型炭材料,2013,28,5,336-341)、纳米金刚石(chemsuschem,2016,9,662-666)、氮化碳(j.mater.chem.a,2014,2,13442-13451)以及其复配制得的纳米金刚石/氮化碳(appl.catal.a:gen.,2019,571,82-88;mater.chem.a,2014,2,13442–13451)催化剂在无水无氧或无水低氧条件下催化乙苯直接脱氢制备苯乙烯的反应中,表现出较好的活性与选择性;到目前为止,人们已经认识到氮原子掺杂到碳基体中可以向非定域π-体系提供额外的电子,并提高碳材料中c=o的化学反应活性。除碳材料外,六方氮化硼(h-bn)因具有热稳定性强、抗氧化性能强和热导率高等特点而被认为是多相催化领域中一种新兴的特色材料,它可以在高放热反应(如f-t合成)中快速消散反应热。据报道,h-bn结构与c3n4相似,是一种用于烷烃脱氢的无金属催化剂,尤其是碳掺杂bn纳米片在丙烷脱氢和乙苯-二氧化碳氧化脱氢(angew.chem.int.ed.,2017,56,8231-8235;j.energy.chem.doi:10.1016/j.jechem.2020.03.027)中的应用,极大地丰富了杂原子掺杂二维无金属材料在催化烷烃脱氢领域的应用。部分材料如商业碳化硼(cn109126843a)也表现出了较高的稳定性,但活性较低。
上述工作极大地拓展了非金属催化剂在乙苯脱氢中的应用,特别是无水条件下直接脱氢制备苯乙烯催化剂的研究。但是由于掺氮碳材料在高温下易于分解,因此大多报道该类催化剂限于在550℃以下应用,使得催化剂的活性、稳定性都不太理想。此外,与无水无氧脱氢相比,无水有氧脱氢易于产生更多的副产物二氧化碳。因此,开发适用于无水条件下,特别是无水无氧条件下,具有耐高温、性能优越、制备工艺简单等优点的非金属脱氢催化剂,仍是目前研究的重点。
申请人所在课题组的前期工作(cn201910739798.4)发现,磷掺杂氮化硼是一类优异的乙苯脱氢非金属催化剂。但目前课题组制备的氮化硼基催化剂,多以粉末为主,粒径尺寸为100~200目,不便于公斤级制备和实际生产中的应用,实用价值受限;而且长期运行,积碳失活的现象依然存在;而且催化剂的选择性还有提升的空间。
技术实现要素:
针对现有烷烃脱氢制烯烃催化剂(本发明也简称为催化剂)存在的催化选择性不理想、容易积碳,长期稳定性不理想等技术问题,本发明第一目的在于,提供一种烷烃脱氢制烯烃催化剂的制备方法,旨在保持良好转化率的同时,改善产物选择性、抗积碳性以及长期稳定性。
本发明第二目的在于,提供所述的制备方法制得的催化剂。
本发明第三目的在于,提供所述的制备方法制得的催化剂的烷烃脱氢方法。
一种烷烃脱氢制烯烃催化剂的制备方法,将能提供b、p、s、n元素的化合物原料在溶剂中进行重结晶,随后将重结晶产物在含氨气气氛内、在大于或等于5℃/min的升温速率下升温至700~1000℃,并进行保温焙烧,制得所述的催化剂;
所述的化合物原料中s:b元素摩尔比为大于或等于10;
p:b元素摩尔比为大于或等于0.5;
n:b的元素摩尔比大于或等于10。
本发明技术方案,创新地采用s、p化合物参与氮化硼的原位制备,并进一步基于原料形态、b:s元素比、b:p元素比以及焙烧机制(例如升温速率以及焙烧温度)的协同控制,能够意外地获得p修饰、且具有特殊微观结构、具有优异催化性能的全新催化剂。
本发明中,在s和p的协同辅助下进行氮化硼原位合成,以及对所述的原料形态、比例和焙烧机制的协同控制是改善制得的催化剂微观结构,并改善其脱氢制烯烃性能的关键。
本发明中,所述的p、s元素需要通过化合物形态引入,如此能够意外地和其他条件协同,从而改善制得的材料的微观结构,并改善其脱氢性能。
作为优选,所述的化合物原料包括硼源、磷源、硫源和氮源,其中,所述的硼源为能够提供硼元素的硼化合物;
所述的磷源为能够提供磷元素的磷化合物;
所述的硫源为能够提供硫元素的硫化合物;
所述的氮源为能够提供氮元素的氮化合物。
本发明中,所述的原料可以是仅提供b、p、s、n中的一种元素的化合物,也可以是能够提供b、p、s、n中的两种及以上的元素的化合物,例如,含有n、s两种元素的化合物既可以用作氮源也可以用作硫源。
优选地,所述的硫源为硫脲、硫代乙硫脲、硫代硫酸铵和硫酸铵中的一种或几种。研究发现,优选的硫源化合物,和其他条件具有意外地协同性,可意外地改善制得的催化剂的微观结构,改善催化剂的性能。进一步优选,所述的硫源为硫脲。优选的化合物能够进一步改善本发明技术方案的协同效果,有助于意外地进一步改善制得的催化剂的性能。
优选地,所述的磷源为羟基乙叉二磷酸、六氯三聚磷腈和磷酸二氢铵中的至少一种。
优选地,所述的硼源选自硼酸和氧化硼中的至少一种。
优选地,所述的氮源为尿素、单氰胺、二氰胺、硫脲和三聚氰胺中的至少一种。
本发明中,对原料中的b、p、s元素的联合控制,能够意外地进一步改善制得的材料的微观结构,改善p修饰杂化,改善其催化性能,特别是有助于改善产物的选择性。
作为优选,化合物原料中,s:b元素摩尔比为10~90:1;进一步优选为16~60:1;更进一步优选为20-60:1:最优选为28-60:1。
作为优选,化合物原料中,p:b元素摩尔比为0.5~2:1;进一步优选为1~2:1;更进一步优选为1~1.4:1。
作为优选,化合物原料中,n:b的元素摩尔比为10~200:1;进一步优选为30~130:1;更进一步优选为40~120:1。
本发明中,可以将能够提供b、s、p和n元素的化合物在溶剂中进行重结晶反应。作为优选,所述的溶剂可以是水;
优选地,重结晶的温度为30~90℃,进一步优选为40~90℃;更进一步优选为50~80℃。
重结晶后可以进行常规的固液分离以及干燥处理,获得所述的产物。
本发明中,将重结晶获得的产物在含氨气氛下进行焙烧,研究发现得益于所述的s、p化合物的辅助,进一步配合升温速率以及温度,能够改善协同性,改善制得的材料的催化性能,特别是有助于改善制得的材料的选择性以及稳定性。
作为优选,升温速率为5~15℃/min;进一步优选为5~10℃/min;进一步优选为5~7.5℃/min。
作为优选,焙烧过程中,所述的含氨气气氛的氨气通入量为5~100ml/min;进一步优选为5~80ml/min;更进一步优选为40~60ml/min。
作为优选,焙烧的温度为700℃~900℃;进一步优选为800℃~900℃。
本发明中,保温焙烧的时间大于或等于2h;优选为2~5h。
本发明优选的制备方法,将硼源、氮源、磷源和硫源的原料水溶液进行重结晶,随后将重结晶产物经干燥、氨气气氛下焙烧得到。所述的磷源为羟基乙叉二磷酸一水合物、六氯三聚磷腈、磷酸二氢铵中的一种或几种复配;所述的硫源为硫脲、硫代乙硫脲、硫酸铵、硫代硫酸铵中的一种或几种;所述的硼源选自硼酸、氧化硼中的一种;所述的氮源选自尿素、单氰胺、二氰二胺、三聚氰胺中的一种或几种。所述的原料液重结晶的温度为40~90℃;所述重结晶产物的真空干燥温度为50~80℃,所述干燥时间为10~20h;所述氨气气氛焙烧时氨气通入量为5~100ml/min,升温速率为5~15℃/min,焙烧温度为700~900℃,焙烧时间为2~5h。
本发明中,还可将所述的催化剂负载在载体上。
本发明中,所述的载体可以是行业内公知的载体,且可以采用行业公知的负载方式进行负载制备。
优选地,所述的载体为三氧化二铝、二氧化硅中的至少一种或者含有二者之一成分的其他载体;
例如,所述的负载的步骤例如为:
将能提供b、p、s和n元素的化合物原料、甲醛以及能够转化为载体的前体原料在溶剂中进行重结晶,随后将重结晶产物进行所述的梯度焙烧,即得。
进一步具体的负载型催化剂(结构化催化剂)的制备过程例如为:
采用甲醛等作为鼓泡介质的自成型的技术。具体为:称取所需质量的氮源物料,加入甲醛溶液,在30-90℃搅拌条件下冷凝回流至澄清。加入所需配比的硼源、磷源、硫源的原料,继续回流1-10小时。将样品转移到容器内,加入一定质量的硅酸钠、硅溶胶或铝溶胶的一种,搅拌均匀,于100-200℃恒温热处理12-36小时。将热处理后的样品取出破碎后过筛,取一定目数,置于管式炉内,通入5-80ml/min的氨气,以进行梯度焙烧,然后自然降温至室温,即得到负载型催化剂。
作为同一发明构思的方案,本发明另一负载型催化剂的制备例如为:将载体和合成的原料混合进行所述的重结晶以及焙烧处理,即得。
本发明还包括所述的制备方法制得的催化剂。所述的催化剂,形态上可以为纳米、微米或毫米级的粉末状,也可以是更大尺寸的、采用自成型等手段进一步获得的结构化形态。
本发明创新地制备方法,其基于化合物s以及p参与bn的原位合成,并基于条件的联合控制,其能够有助于p的修饰以及修饰形态的控制,此外,还能够具有化合物s的作用,对材料的微观结构进行控制,能够意外地获得具有特殊化学以及物理结构的新材料。
本发明还公开了一种烷烃脱氢制烯的方法,将烷烃原料和所述制备方法制得的催化剂接触,进行脱氢反应,制得相应的烯烃产物。
本发明中,得益于所述的催化剂的使用,可实现烷烃的直接脱氢或者氧化脱氢,高效获得相应的烯烃,并具有良好的转化率、产物选择性及稳定性。
作为优选,所述的烷烃原料为具有式1结构式的化合物:
式1
其中,r1~r4独自为h、c1~c10的烷基、c3~c10的环烷基或芳基;或者,r1与r4相互环合形成环基;
所述的烷基、环烷基、环基或芳基上允许带有取代基,分别为取代烷基、取代环烷基、取代环基、取代芳基;其中,所述的取代基为c1~c6的烷基、c1~c6的烷氧基、卤素、苯基、硝基、三氟甲基中的至少一种取代基。
本发明所述的烷基为直链或者支链烷基。所述的环烷基为三元~六元的单环、螺环或桥环。所述的芳基为苯环、五元杂环芳基、六元杂环芳基、或者由苯环、五元杂环芳基、六元杂环芳基中的两个及以上芳香环并合形成的稠环。
此外,所述的r1与r4相互环合形成环基,所述的环基例如为五元或者六元的环。例如,环合后的烷烃原料结构例如为
本发明中,在所述的催化剂催化下,将式1直接或氧化脱氢,获得式2的烯烃产物
优选地,所述的烷烃原料为具有式1-1结构的化合物;
式1-1中,r1为h、c1~c6的烷基、苯基或者取代苯基;所述的取代苯基的苯环上含有c1~c6的烷基、c1~c6的烷氧基、卤素、苯基、硝基、三氟甲基中的至少一种取代基。
在本发明所述的催化剂催化下,可将式1-1直接催化制成相应的端烯烃产物(r1-=;式2-1)。
作为优选,脱氢反应在无水条件下进行;或者在含弱氧气氛下进行。含弱氧气氛的氧含量例如不高于10vol.%;例如可以为1~5vol.%。
优选地,脱氢反应在有氧气氛或者无氧气氛下进行;
优选地,脱氢反应的温度为500~700℃;优选为550~650℃;进一步优选为600~650℃。
本发明中,所述的脱氢反应可以基于现有的反应设备实现,例如,可以将其填充至反应器中,并进行气固相催化反应。
原理:
目前,对于非金属催化剂在烃类脱氢反应中的活性位,大多数研究者认为是存在于碳材料和氮化硼材料表面的c=o基团或者b-oh基团。本发明所制备的催化剂,从后边的实施例、对比例,并利用xps对表面基团、利用氮气物理吸附对孔结构的分析结果可知,n3p-oh、n2p=o及n-b-o等基团均可以作为本反应的活性基团;而且,催化剂的微观结构对选择性和稳定性影响显著;通过在氮化硼原位合成阶段引入硫元素以及磷元素化合物,基于硫化合物、磷化合物以及条件的协同,能够改善p的修饰,并同时能够调控材料的微观结构,进而有助于改善制得的材料的催化性能。
有益效果:
1、本发明创新在硫-磷化合物协同下进行氮化硼的原位合成,并进一步基于s、p物料形态、含量以及焙烧机制的协同下,能够改善p对bn的修饰,此外,还有助于调控微观结构,进而在保持良好转化率的同时,显著改善产物的选择性,改善积碳问题,改善催化剂的稳定性。本发明以典型的乙苯脱氢为例,单位催化剂、单位时间内实现的苯乙烯生成量可达24.33mmol/(g·h),苯乙烯选择性大于97%,稳定运行100小时以上。对于具有实际意义的工业化反应,具有重要的经济价值、环保价值和社会价值。
2、本发明进一步研究发现,传质状况对本反应催化剂的选择性和稳定性影响显著。通过控制所述催化剂制备过程的硫源种类、b/s比、以及焙烧温度、焙烧时间,以及结构化的做法,有助于调控催化剂的孔结构,进一步改善烷烃脱氢的选择性和稳定性。
3、本发明进一步研究发现,n3p-oh、n2p=o及n-b-o等基团均可以作为本反应的活性基团,从而为全新催化剂的设计提供理论指导。
4、结构化催化剂具有更好的机械强度和成型性,可以满足不同条件的生产技术需求。
5、本发明所述的催化剂与目前工业中使用的含钾氧化铁催化剂相比,具有耐高温、抗积碳能力更强、制备工艺简单、无金属污染等优点,具有非常好的工业化应用前景。
附图说明
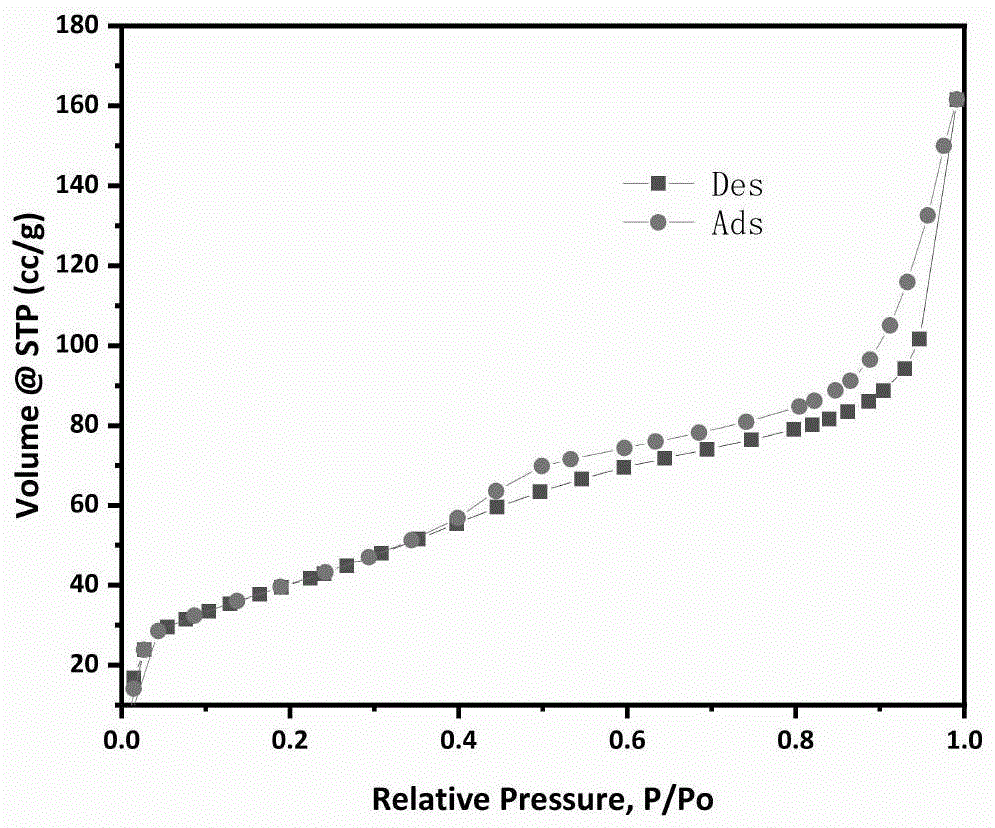
附图1:对比例1的催化剂的氮气吸附-脱附曲线
附图2:实施例1的催化剂的氮气吸附-脱附曲线
附图3:实施例1的催化剂的xps图
附图4:实施例1的催化剂的tem图
附图5:实施例11的催化剂的sem图
具体实施方式
以下结合实施例详述本发明。
实施例1
称取一定量的硼酸(硼源,0.4g)、硫脲(硫源和氮源)、羟基乙叉二膦酸(磷源),原料中,b:n:s:p的摩尔比为1:90:48:1.2),加入40ml蒸馏水,搅拌溶解,然后将装有含前驱物溶液的烧杯在80℃油浴、300rpm搅拌条件下进行重结晶操作,至无明显水分,然后将得到的白色重结晶产物转入50℃真空干燥箱中进一步干燥12h。将干燥后的重结晶产物研磨成粉,装到刚玉方舟中,置于管式炉内,通入60ml/min的氨气提供焙烧气氛,以5℃/min的升温速率自室温升温至800℃焙烧3h,然后在氨气保护下(氨气的流量为20ml/min)自然降温至室温,即得到催化剂,记:bnsp-硫脲/hedp-48/1.2。制得的材料的氮气吸附-脱附曲线图2;xps图见图3;tem图见图4。
从图1、图2对比可以看出,制得的材料的氮气吸附-脱附曲线出现h3型回滞环,即存在狭缝孔结构,比表面积为101.13m2/g,该材料的比表面积比前期工作中bnp-1.2(对比例1)的比表面积(140~260m2/g)小,结合孔径分析,材料的平均孔径为13.39nm,远大于bnp-1.2的孔径(3nm),因此比表面积低于bnp-1.2。此外,图3的xps测试发现,s的原子百分比低于仪器的检测线,即s很难进入到bn骨架或表面,因此可推断s源的作用主要是影响了材料热处理过程中催化剂微观结构的形成。
催化剂性能的测试方法为:将催化剂50mg,加入2ml粒度为40~60目的石英砂稀释,装入φ8mm的固定床石英反应管中,催化剂床层两端以少量石英棉封堵。通入20ml/min的氮气提供惰性气体气氛。氮气保护下,以4℃/min的速率升温到600℃,稳定30min对催化剂进行预活化,然后以20ml/min的流速通入乙苯体积分数为2.8%的混合原料气,进行连续反应。反应产物用5℃的乙醇收集,通过岛津gc-2010plus气相色谱仪分析其组成,色谱柱型号为rtx-5,检测器为fid。初始乙苯转化率77.43%,苯乙烯选择性97.25%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为22.72mmol/(g·h),可以稳定40小时以上。
实施例2
与实施例1相比,区别仅在于,改变了硫源的成分,分别采用硫代硫酸铵、硫酸铵进行研究,其中,以硫代硫酸铵作硫源时,以尿素做补充氮源,以控制b:n的比例保持不变,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表1所示:
表1:
实施例3
与实施例1相比,区别仅在于,改变硫源的摩尔比,分别为b:s的摩尔比分别为1:16、1:20、1:24、1:28,并以尿素做补充氮源,以保持b:n不变,另外,b:s的摩尔比为1:60案例中,未额外添加尿素作为氮源;其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表2所示:
表2
a:未额外添加尿素。
实施例4
与实施例1相比,区别仅在于,改变了磷源的成分,分别采用磷酸二氢铵、六氯三聚磷腈进行研究,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表3所示:
表3
实施例5
与实施例1相比,区别仅在于,改变磷源的摩尔比,分别为b:p的摩尔比分别为1:1.0、1:1.4,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表4所示:
表4
由实施例5以及我们的前期工作可以看出,b:p摩尔比的大小决定了制备得到的材料中磷元素原子百分比以及活性位点n2p=o和n3p-oh的相对比例。当b:p≤1:1.4时,制备得到的材料中磷元素原子百分比增加,n2p=o和n3p-oh的相对比例逐渐降低。结合dft理论计算,n2p=o促进了乙苯中c-h键的断裂,n3p-oh降低了断裂后产生的h*结合解吸的能垒,二者协同作用下实现乙苯直接脱氢生成苯乙烯的反应。当b:p>1:1.4时,由于hedp的使用在引入p的同时会引入大量的c和o,导致所制备的材料中磷元素的原子百分比降低,所以催化性能较差。综合考虑,优选的b:p摩尔比为1:0.9~1.4。
实施例6
与实施例1相比,改变升温速率(升温至焙烧温度的速率),分别为7.5℃/min、10℃/min,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表5所示:
表5
由实施例1和6可以看出,在大于或等于5℃/min的升温速率下特别是优选的5~7.5℃/min可以获得性能优异的催化性能。
实施例7
与实施例1相比,改变焙烧的温度,分别为600℃、700℃、900℃,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表6所示:
表6
研究发现,在大于或等于600℃的温度,特别是优选的800~900℃,可以获得良好的催化性能。
实施例8
和实施例2中硫代硫酸铵作为硫源相比,区别仅在于,n源的补充分别为单氰胺、二氰二胺,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表7所示:
表7
实施例9
与实施例1相比,同时改变氮气流量、升温速率和焙烧的温度(80ml/min,10℃/min,900℃)。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率63.09%,苯乙烯选择性97.33%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为18.53mmol/(g·h),可以稳定40小时以上。
实施例10
称取市售二腈二胺8.4g,加入10ml市售37%浓度甲醛溶液,置于50ml三颈烧瓶内,在85℃油浴、400rpm条件下冷凝回流至澄清后,在同样条件下继续冷凝回流1h。
称取一定量市售硼酸(0.4g),硫脲,羟基乙叉二膦酸(原料中,b:n:s:p的摩尔比为1:90:48:1.2),加入三颈烧瓶中,继续在85℃油浴、400rpm条件下冷凝回流至澄清后,在同样条件下继续冷凝回流1h。
将样品转移到试管内,称取市售硅酸钠1.5g加入试管,搅拌均匀,盖上有小孔的塞子,放入180℃烘箱中恒温热处理12h。
将热处理后的样品取出砸碎后过筛,取20至40目样品,装到刚玉方舟中,置于管式炉内,通入60ml/min的氨气提供焙烧气氛,以5℃/min的升温速率自室温升温至800℃焙烧3h,然后在氨气保护下自然降温至室温,即得到自成型的硫修饰的磷掺杂氮化硼催化剂,记:bnsp-硫脲/hedp-48/1.2-自成型。
加入2ml粒度为40~60目的bnsp-硫脲/hedp-48/1.2-自成型,采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率78.31%,苯乙烯选择性97.39%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为23.01mmol/(g·h),可以稳定100小时以上。
实施例11(与实施例1相比,负载法制备的结构化催化剂)
与实施例1相比,将20-40目的活性氧化铝颗粒加入到硼酸、硫脲、羟基乙叉二膦酸一水合物的混合溶液中,其他步骤不变。催化剂记为:bnp@氧化铝/hedp-48/1.2。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率82.50%,苯乙烯选择性97.72%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为24.33mmol/(g·h),可以稳定100小时以上。
实施例12(氧化脱氢)
将实施例1的催化剂,应用于弱氧氛围下的氧化脱氢。与实施例1相比,催化剂的性能测试方法中,20ml/min的氮气改为含有氧气3%(体积百分数)的氮氧混合气,测得催化剂的性能为:初始乙苯转化率78.35%,苯乙烯选择性94.60%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为22.36mmol/(g·h),可以稳定100小时以上。
实施例13
和实施例1相比,区别仅在于,脱氢反应过程的温度分别为550℃、575℃、625℃,其他参数以及操作均同实施例1。
采用实施例1中的催化剂性能测试方法,测得催化剂的性能见表7所示:
表7
对比例1
和实施例1相比,主要仅在于,未添加硫源,氮源由尿素代替,具体步骤同申请号为201910739798.4的实施例1;催化剂记为bnp1.2。
该对比例的催化效果为:初始乙苯转化率62.42%,苯乙烯选择性93.54%,对应的单位催化剂上单位时间内实现的乙苯转化量为17.62mmol/(g·h),可以稳定20小时以上。其效果显著差于实施例1,且产物的选择性差于本发明实施方案。
对比例2
与实施例1相比,区别仅在于,采用单质硫(升华硫,添加摩尔量同实施例1;n源由尿素提供,且n源的摩尔量同实施例1),其他条件均同实施例1,催化剂记为:bnsp-升华硫/hedp-48/1.2。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率22.36%,苯乙烯选择性93.71%。对应的单位催化剂上单位时间内实现的苯乙烯生成量为6.32mmol/(g·h)。
对比例3
与实施例1相比,区别仅在于,不添加磷源。催化剂记为:bns-48。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:29.73%,苯乙烯选择性97.68%。对应的单位催化剂上单位时间内实现的苯乙烯生成量为8.76mmol/(g·h),可以稳定20小时以上。其效果显著差于实施例1,主要是产物的转化率差于本发明实施方案。
对比例4
与实施例1相比,区别仅在于,磷源为磷单质(红磷,添加摩尔量同实施例1)。催化剂记为:bnsp-硫脲/p-48-1.2。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率20.04%,苯乙烯选择性90.68%。对应的单位催化剂上单位时间内实现的苯乙烯生成量为5.48mmol/(g·h)。
对比例5
与实施例1、实施例5相比,区别仅在于,硼元素与磷元素的摩尔比为1:0.2),催化剂记为:bnsp-硫脲/hedp-48/0.2。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率28.42%,苯乙烯选择性95.31%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为8.17mmol/(g·h)。
对比例6
与实施例1、实施例6相比,区别仅在于,升温速率为1℃/min。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率46.59%,苯乙烯选择性92.42%。对应的单位催化剂上单位时间内实现的苯乙烯生成量为12.99mmol/(g·h),可以稳定20小时以上。
对比例7
与实施例1、实施例7相比,区别仅在于,焙烧温度为1100℃。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率50.33%,苯乙烯选择性83.03%。对应的单位催化剂上单位时间内实现的苯乙烯生成量为12.61mmol/(g·h),可以稳定20小时以上。
对比例8
探讨bn和硫源和p源物理混合:
与实施例1相比,区别主要在于,先不加磷源和硫源,直接合成bn(合成方式同实施例1,例如,采用b在氨气气氛下进行烧结);随后再将bn和硫源(硫脲)和p源按比例混合,进一步在氮气保护气氛下热处理。采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率18.51%,苯乙烯选择性85.47%。对应的单位催化剂上单位时间内实现的苯乙烯生成量为4.77mmol/(g·h),可以稳定20小时以上。
对比例9(空白载体石英砂)
量取2ml粒度为40~60目的石英砂,采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率8.82%,苯乙烯选择性58.33%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为1.55mmol/(g·h)。
对比例10(空白载体硅酸钠)
量取2ml硅酸钠,采用实施例1中的催化剂性能测试方法,测得催化剂的性能为:初始乙苯转化率7.80%,苯乙烯选择性62.45%,对应的单位催化剂上单位时间内实现的苯乙烯生成量为1.47mmol/(g·h)。
分析:
从实施例、对比例的结果来看,(1)硫元素的掺杂,可以显著提高磷氮硼催化剂对苯乙烯的选择性。(2)硫源、磷源的共同加入,是获得高性能催化剂的关键之一。(3)硫源类型、磷源类型,及添加量,是获得高性能催化剂的关键之二。(4)升温制度是获得高性能催化剂的关键之三。从附图可以看出,催化剂是明显的多孔材料。硫元素主要影响着催化剂合成过程中孔结构的形成。使得催化剂孔结构更加合理,动力学上有利于中间体的传递和转化,避免副反应的发生。
- 还没有人留言评论。精彩留言会获得点赞!