一种赤灵芝药材的质量评价方法与流程
本发明属于药材的质量控制领域,特别涉及一种赤灵芝药材的质量控制方法。
背景技术:
灵芝在我国已有上千年的历史,早在《神农本草经》中就有记载,灵芝固本培元,久食不老,轻身延年。
然而,灵芝作为一种真菌类药材,品种繁多,不同品种的灵芝化学成分差异巨大,即使同为赤灵芝,又分为很多亚种,灵芝的不同部位子实体与孢子粉成分也完全不同,菌丝体也与子实体在成分上有很大差异。中药的功效是建立在其所含有的化学物质的组成和含量稳定的基础上的,如果化学组成和含量差异很大,那么其功效必然会产生很大的不确定性。同时,以化学组成和含量差异巨大的灵芝药材为原料生产的灵芝产品也必然无法做到质量稳定可靠,疗效确切。
目前灵芝通用的评价方法是2015版中国药典提出的,该质量标准专属性差。其薄层鉴别方法无法区分不同品种的灵芝,其含量测定方法不足以评价灵芝质量的的优劣。
本领域内有学者用液相色谱法,以灵芝中不同灵芝酸化学对照品为对照,检测灵芝药材中灵芝三萜含量。但这种方法由于对照品过于稀有昂贵,同时单一一种灵芝酸的含量无法代表灵芝中全部灵芝酸的含量。因此难于应用于生产和流通领域。
技术实现要素:
本发明为弥补现有技术中存在的一些不足,提供一种赤灵芝药材的质量评价方法,利用该方法不仅可更加准确、全面、便捷的评价赤灵芝质量优劣,还可降低检测成本。
本发明为达到其目的,采用的技术方案如下:
一种赤灵芝药材的质量评价方法,包括如下步骤:以赤灵芝提取物对照品为对照,用液相色谱法在210nm-280nm检测波长范围内的任意波长下测定待测赤灵芝药材样本,在得到的液相色谱图中对保留时间为15~65分钟区间的所有紫外吸收峰面积求和,并与赤灵芝提取物对照品中的灵芝酸A所对应的紫外吸收峰面积相比较,计算待测赤灵芝药材样本中总灵芝三萜含量,以总灵芝三萜含量作为评价赤灵芝药材质量优劣的质量指标。
进一步的,保留时间为15~65分钟区间内的任意单一紫外吸收峰面积在该区间所有紫外吸收峰面积之和的占比≤30%。
优选的,还包括如下步骤:以赤灵芝提取物对照品为对照,用薄层色谱方法对待测赤灵芝药材的真伪进行薄层鉴别。
优选的,所述赤灵芝提取物对照品中含有如下质量百分含量的各组分:灵芝酸A为0.3%-0.5%,灵芝酸B为0.15%-0.4%,灵芝酸C2为0.3%-0.5%,灵芝酸G为0.25%-0.5%。
进一步的,所述赤灵芝提取物对照品的制备包括如下步骤:对赤灵芝进行提取以获得赤灵芝提取物,将不同的赤灵芝提取物进行相互勾兑以获得赤灵芝提取物对照品,对赤灵芝提取物对照品中的灵芝酸A的含量进行标定。
优选的,所述赤灵芝提取物的制备包括如下步骤:将赤灵芝粉碎,加入溶剂提取,对提取液进行过滤,收集滤液,对滤液进行减压浓缩,向浓缩物中加入辅料并继续减压浓缩至干;进一步优选的,所述提取采用加热回流提取、超声提取、微波提取、渗漉提取或闪式提取;进一步优选的,所述辅料为糊精、微粉硅胶、淀粉、羧甲基纤维素中的至少一种;进一步优选的,所述辅料和所述浓缩物的质量比为0.5∶1~3∶1;进一步优选的,所述溶剂为甲醇、乙醇、体积浓度为70%以上的甲醇水溶液、体积浓度为70%以上的乙醇水溶液中的至少一种,其中,最优选100%甲醇或95%乙醇水溶液。
作为一种优选实施方式,所述液相色谱法的色谱条件包括:色谱柱选择反相色谱柱,流动相包括流动相A和流动相B,流动相A为三氟乙酸、甲酸、乙酸、磷酸中的至少一种与甲醇、乙腈中的至少一种的混合溶液,流动相B为三氟乙酸、甲酸、乙酸、磷酸中的至少一种的水溶液。
进一步优选的,所述流动相A为0.05~0.15%(质量)三氟乙酸乙腈溶液,流动相B为0.05~0.15%(质量)三氟乙酸水溶液;采用梯度洗脱,在0~120min,流动相A的体积比例在20%~100%之间逐渐增长。
更进一步优选的,流动相的梯度洗脱程序为:A+B=100%(体积),在0-35min,流动相A维持在20-30%,流动相B维持在70-80%;在35-90min,流动相A维持在60-70%,流动相B维持在30-40%;在90-120min,流动相A维持在100%,流动相B维持在0%。
作为一种具体实施方式,色谱条件还包括:流速为1.2mL/min;柱温为25℃;检测波长范围为210nm-280nm,其中优选波长254nm。
优选的,所述薄层色谱方法采用的薄层板为硅胶薄层板,展开剂采用体积比为12~17∶12~17∶0.5~1.5∶0.1的甲苯、乙酸乙酯、甲醇、甲酸,显色剂采用质量浓度10%-50%的硫酸乙醇溶液。展开方法可以是一次展开也可以是多次展开。
优选的,待测的赤灵芝药材按照如下步骤进行前处理:将药材用10-20倍质量的甲醇超声15-45min,过滤,滤渣再次用10-20倍质量的甲醇超声15-45min,过滤,合并滤液,水浴蒸干滤液,用甲醇溶解并定容;赤灵芝提取物对照品用甲醇溶解并定容后用于对照。
对赤灵芝药材进行薄层色谱鉴别,其鉴别标准为,赤灵芝药材的薄层图谱需与赤灵芝提取物对照品的薄层图谱在相应的位置上,显相同颜色的荧光斑点。
对赤灵芝药材进行液相色谱检测,根据赤灵芝药材中总灵芝三萜的含量进行质量分级,含量大于13mg/g的药材为特级品;总灵芝三萜含量在8-13mg/g范围的药材为一级品;总灵芝三萜含量在5-8mg/g范围的药材为二级品;总灵芝三萜含量在1-5mg/g范围的药材为三级品;总灵芝三萜小于1mg/g的药材为等外品。
本发明提供的技术方案具有如下有益效果:
1、本申请发明人在对灵芝大量的质量研究的基础上,建立了赤灵芝药材质量控制方法,在优选方案中,通过结合薄层色谱鉴定和液相色谱检测,可以准确的鉴别赤灵芝的真伪,并评价赤灵芝的优劣。
2、本发明的薄层色谱鉴别保证了赤灵芝在品种上的一致性。液相色谱的含量测定通过采用本发明优选的流动相和梯度洗脱方法得到的液相色谱图中对保留时间在15-65分钟区间的所有紫外吸收峰面积求和,以赤灵芝提取物对照品中的灵芝酸A为对照,计算样品赤灵芝中灵芝总三萜含量,保证了赤灵芝药材中的有效成分灵芝三萜含量检测的准确性。
本发明人研究了采用本液相色谱方法测定赤灵芝药材的液相色谱图,并对保留时间在15-65分钟区间的主要组分进行全波长扫描,发现该区间内的主要成份的紫外吸收色谱非常相似,通过计算发现这些组分具有相似的摩尔吸光系数,因此可以用其中一种代表性组分为对照对该区间内的主要吸收峰进行峰面积求和,计算赤灵芝药材中总灵芝三萜含量。
灵芝酸A是一种灵芝三萜的代表性成分,在灵芝中含量较高,该化学对照品相对易得。因此本发明选定以灵芝酸A的含量为基础,折算赤灵芝药材中的总灵芝三萜含量。
目前通用的方法中灵芝三萜的含量是以熊果酸或齐墩果酸为对照品通过紫外分光光度法测定的。熊果酸或齐墩果酸的化学结构与灵芝酸A的差别较大,同时用紫外分光光度法测定灵芝三萜专属性差,因此该方法在实践中无法确认赤灵芝药材质量的优劣。本发明很好的解决了上述问题。本发明用赤灵芝提取物对照品代替熊果酸或齐墩果酸对照品,并用液相色谱法代替紫外分光光度法,以灵芝酸A为基础计算赤灵芝药材中的灵芝总三萜,大大提高了检验的精度和专属性。
3、以赤灵芝提取物对照品为对照,进行薄层色谱的鉴别,而非使用单一灵芝酸的化学对照品(或几种灵芝酸的化学对照品的组合)为对照或对照药材为对照进行鉴别,具有显著优越性。与使用单一灵芝酸的化学对照品(或几种灵芝酸的化学对照品的组合)为对照相比,使用提取物对照品,可以呈现更加丰富的图谱信息,便于不同灵芝品种间的鉴别。与使用对照药材为对照进行鉴别相比,使用提取物对照品鉴别具有更好的稳定性和更强的通用性。即使是对照药材,也会受到自然环境的影响产生较大的批次差异,而提取物对照品由于可以实现生产工艺的调控和原料的勾兑,并且经过了严格标定,因此可以更好的保证批次间的均一性。除此之外,赤灵芝提取物对照品只需要简单配制就可直接使用,不像对照药材使用前需要提取。因此赤灵芝提取物对照品使用更加简洁方便。
4、优选以赤灵芝提取物对照品为对照,而非以单一灵芝酸的化学对照品或几种灵芝酸的化学对照品的组合为对照,标定赤灵芝药材的三萜含量,该方法具有显著优越性。
现有的灵芝三萜化学对照品提取纯化工艺复杂,成本高昂,难于在生产实践中推广。而本发明的赤灵芝提取物对照品制备工艺简单,重复性好,能够同时满足定性、定量检测灵芝中灵芝酸的要求。
附图说明
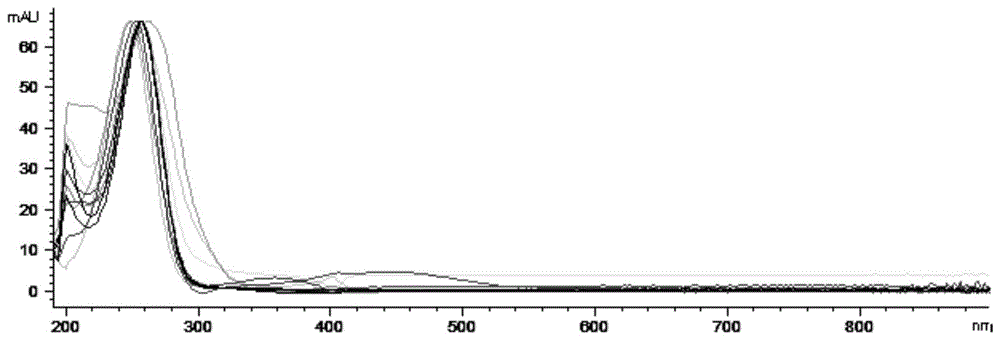
图1表2-4主要组分紫外全波长扫描图的叠加图;
图2是采用中国药典(2015版)中灵芝薄层鉴别的方法得到的薄层色谱鉴别图谱;
图3是采用本发明的灵芝薄层鉴别的方法得到的薄层色谱鉴别图谱;
图4是采用中国药典(2015版)方法检测赤灵芝药材三萜及甾醇含量的含量测定图;
图5是用本发明的赤灵芝药材的检测方法测定赤灵芝药材灵芝三萜含量的含量测定图;
图6是用本发明的赤灵芝药材的检测方法测定赤灵芝药材的液相色谱图;
图7是用本发明的灵芝薄层鉴别方法检测赤灵芝药材的薄层色谱鉴别图谱;
图8赤灵芝药材含量测定图。
具体实施方式
本发明提供一种赤灵芝药材的质量评价方法,主要包括如下步骤:以赤灵芝提取物对照品为对照,在210nm-280nm的检测波长下测定待测赤灵芝药材样本,对保留时间为15~65分钟区间的所有紫外吸收峰面积求和,并与赤灵芝提取物对照品中的灵芝酸A所对应的紫外吸收峰面积相比较,计算待测赤灵芝药材样本中总灵芝三萜含量,以总灵芝三萜含量作为评价赤灵芝药材质量优劣的质量指标。优选的,进一步包括如下步骤:通过薄层色谱法对赤灵芝药材进行薄层鉴别。通过薄层色谱鉴别保证了赤灵芝在品种上的一致性。采用本发明的方法能更准确的测定灵芝药材中的有效成分灵芝三萜的含量,从而利于辨别不同药材质量的好坏。
作为优选方案,液相色谱检测和薄层色谱鉴别时,以赤灵芝提取物对照品为对照;赤灵芝提取物对照品的质量标准为含有如下质量百分含量的各组分:灵芝酸A为0.3%-0.5%,灵芝酸B为0.15%-0.4%,灵芝酸C2为0.3%-0.5%,灵芝酸G为0.25%-0.5%。
进一步优选的,赤灵芝提取物对照品的制备包括如下步骤:对赤灵芝进行提取以获得赤灵芝提取物,将不同的赤灵芝提取物进行相互勾兑以获得赤灵芝提取物对照品,对赤灵芝提取物对照品中的灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G的含量进行标定。所述不同的赤灵芝提取物为不同批次提取获得的赤灵芝提取物,或两种以上赤灵芝的提取物。优选的,所述赤灵芝提取物对照品为两种以上赤灵芝的提取物勾兑而成,从而可以使制得的提取物对照品的条带更丰富,不同成分的含量也更加均匀,从而得到更为优质的提取物对照品。
更进一步优选的,所述赤灵芝提取物的制备包括如下步骤:将赤灵芝粉碎,加入溶剂提取,对提取液进行过滤,收集滤液,对滤液进行减压浓缩,向浓缩物中加入辅料并继续减压浓缩至干;优选的,所述提取采用加热回流提取、超声提取、微波提取、渗漉提取或闪式提取;优选的,所述辅料为糊精、微粉硅胶、淀粉、羧甲基纤维素中的至少一种;优选的,所述辅料和所述浓缩物的质量比为0.5∶1~3∶1;优选的,所述溶剂为甲醇、乙醇、体积浓度为70%以上的甲醇水溶液、体积浓度为70%以上的乙醇水溶液中的至少一种。最优选100%甲醇或95%乙醇水溶液。
作为一种优选方案,所述液相色谱法的色谱条件包括:色谱柱选择反相色谱柱,流动相包括流动相A和流动相B,流动相A为三氟乙酸、甲酸、乙酸、磷酸中的至少一种与甲醇、乙腈中的至少一种的混合溶液,流动相B为三氟乙酸、甲酸、乙酸、磷酸中的至少一种的水溶液。作为进一步优选方案,所述流动相A为0.05~0.15%(质量)三氟乙酸乙腈溶液,流动相B为0.05~0.15%(质量)三氟乙酸水溶液;采用梯度洗脱,在0~120min,流动相A的体积比例在20%~100%之间逐渐增长。作为更进一步优选方案,流动相的梯度洗脱程序为:A+B=100%(体积),在0-35min,流动相A维持在20-30%,流动相B维持在70-80%;在35-90min,流动相A维持在60-70%,流动相B维持在30-40%;在90-120min,流动相A维持在100%,流动相B维持在0%。
作为一种优选方案,所述薄层色谱鉴别方法采用的薄层板为硅胶薄层板,展开剂采用体积比为12~17∶12~17∶0.5~1.5∶0.1的甲苯、乙酸乙酯、甲醇、甲酸,显色剂采用质量浓度10%-50%的硫酸乙醇溶液。展开方法可以是一次展开也可以是多次展开。
下面结合附图和具体实施例对本发明的技术方案做进一步说明。
实施例1赤灵芝提取物对照品的制备
1.1、实验概述
赤灵芝提取物制备所用溶剂为甲醇,提取方式为加热回流。
1.2、实验方法
将2种经菌种鉴定确认为赤灵芝的药材分别制备成赤灵芝提取物,制备方法如下:称取赤灵芝药材2kg粉碎,加入8倍量的甲醇16L,加热回流提取两次,每次1小时(液体沸腾开始计时),滤过,收集滤液,在65℃下减压浓缩至5L,各加入辅料微粉硅胶120g,继续浓缩至干,得到赤灵芝提取物各200g,分别编号为1号赤灵芝提取物、2号赤灵芝提取物。
称取1号赤灵芝提取物200g,2号赤灵芝提取物200g,混合均匀,加入甲醇4L,搅拌使充分溶解混匀,浓缩至干,得到勾兑后的赤灵芝提取物对照品。
实施例2标定赤灵芝提取物对照品
2.1实验概述
以灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G为对照品,用液相色谱法标定赤灵芝提取物对照品中灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G的含量。标定方法如下:
2.2试剂
灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G均购自上海源叶生物技术有限公司
2.3赤灵芝提取物对照品及灵芝酸对照品的样品制备
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
分别称取灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G对照品5mg,用甲醇溶解定容至5ml容量瓶中。过0.22um微孔滤膜,即得。
2.4液相色谱条件
色谱柱: TSK gel ODS-80Tm(5μm 150*4.6mm)
流动相: 按下表比例,梯度洗脱
流速:1.2mL/min;柱温:25℃;检测波长:254nm进样量:10μl
2.5实验结果:
用灵芝酸化学对照品标定赤灵芝提取物对照品中的灵芝酸含量,含量数据如下表1。该方法中灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G的分离度均大于0.9,理论塔板数均大于6000,RSD均小于1%,因此该方法可以用于标定赤灵芝提取物对照品。用灵芝酸化学对照品标定赤灵芝提取物对照品含量,赤灵芝提取物对照品的灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G含量为:灵芝酸A:0.4108%,灵芝酸B:0.2286%,灵芝酸C2:0.3402%,灵芝酸G:0.3168%。
表1
实施例3赤灵芝提取物对照品主要组分(保留时间15-65min)的紫外吸收分析
3.1实验概述
用液相色谱方法测定赤灵芝提取物对照品,分析图谱中液相色谱保留时间15-65min的峰的紫外吸收。
3.2样品制备
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
3.3液相色谱条件
色谱柱: TSK gel ODS-80Tm(5μm 150*4.6mm)
流动相: 按下表比例,梯度洗脱
流速:1.2mL/min;柱温:25℃;检测波长:254nm进样量:10μl
3.4实验结果:
用液相色谱方法分析赤灵芝提取物对照品,对液相图谱中保留时间为15-65min的主要组分进行全波长扫描,其数据见表2-表4及图1。图1为表2-4中各组分紫外全波长扫描图的叠加图。
由图1可以看出液相图谱中保留时间为15-65min的主要组分的全波长扫描图非常相似,说明这些主要组分具有相似的化学结构,属于同类的化合物。又因为灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G四种已知的灵芝三萜成分的保留时间均落在该区间,由此推断该区间的组分主要为灵芝三萜类化合物。因此可以以一种灵芝酸的代表性组分为基础,对该区间所有主要组分的紫外吸收峰进行积分求和,计算赤灵芝的灵芝总三萜含量。
从表2-表4及图1可以看出:在选取的10个主要组分中,有7个组分的最大吸收波长为254nm(包括灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G),有2个组分的紫外最大吸收波长是246nm,另有1个组分的紫外最大吸收波长为262nm。因此选取254nm作为检测灵芝总三萜最优的检测波长。
表2
表3
表4
实施例4中国药典灵芝药材薄层色谱检测方法的局限性及方法优化
4.1.实验概述
用中国药典中的薄层色谱鉴别方法与优化后的薄层色谱鉴别方法进行对比,两种薄层色谱鉴别方法如下:
4.2.中国药典薄层色谱鉴别方法
4.2.1.样品制备方法
分别称取2g灵芝药材样品及2g灵芝孢子粉,加入30ml乙醇加热回流30min,过滤,水浴蒸干滤液,残渣加甲醇2ml溶解。即得。
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。即得。
4.2.2.中国药典薄层色谱色谱条件:
薄层板: 硅胶薄层板
点样: 点样量2μl,
展开剂: 石油醚(60-90℃)∶甲酸乙酯∶甲酸=15∶5∶1(上层溶液)
检视: 展开后薄层板放置在空气中干燥,在紫外灯光UV366nm下检视。
4.3.优化后的薄层色谱实验方法
4.3.1.样品制备方法
分别称取2g灵芝药材样品及2g灵芝孢子粉,加入30ml甲醇超声(150w,44khz)30min,过滤,滤渣加入30ml甲醇超声30min,过滤,滤纸用少量甲醇潤洗,水浴蒸干滤液,用甲醇溶解并定容至2ml容量瓶中。即得。
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。即得。
4.3.2.优化后的薄层色谱条件:
薄层板: 硅胶薄层板
点样: 点样量2μl
展开剂: 甲苯∶乙酸乙酯∶甲醇∶甲酸=15∶15∶1.0∶0.1
检视: 喷加10%硫酸乙醇试剂,烘干,在紫外灯光UV366nm下检视。
4.3.3.实验结果:
薄层色谱检测的图谱见图2和图3,从图2和图3的对比可以看出中国药典的薄层色谱鉴别方法图谱信息量少,各样品在图谱上只显示一条谱带,各样品谱带间无明显差异,无法区分不同品种灵芝。优化后的薄层色谱鉴别方法图谱信息丰富,各样品在图谱上显示多条谱带,且谱带的粗细、颜色、深浅均有明显差异,能够反映各样品的品种特点和质量差异。例如:图3中可以明显发现4号样品、5号样品、6号样品的谱带特征与赤灵芝提取物对照品的谱带不同,事实上这3个样品确实不是赤灵芝品种。采用本发明的薄层色谱检测,通过比较所检测的灵芝药材和赤灵芝提取物对照品的薄层图谱在相应的位置上是否具有显相同颜色的荧光斑点,可以达到鉴别目的。可见,以赤灵芝提取物对照品为对照使用优化后的薄层色谱鉴别方法能更好的鉴别赤灵芝药材。
图2和图3中的标号分别对应如下样品:1.赤灵芝提取物对照品、2.赤灵芝1、3.赤灵芝2、4.扛四灵芝、5.树舌灵芝、6.紫芝、7.灵芝孢子粉。
实施例5用赤灵芝提取物对照品和灵芝酸化学对照品标定同一赤灵芝药材灵芝酸含量
5.1实验概述
用赤灵芝提取物对照品和灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G四个化学对照品及用液相色谱方法标定同一种赤灵芝药材,确认用赤灵芝提取物对照品标定赤灵芝药材中灵芝酸含量的准确性。其方法如下:
5.2试剂
赤灵芝提取物对照品为实施例1制备,赤灵芝药材,灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G均购自上海源叶生物技术有限公司。
5.3样品制备
称取2g赤灵芝药材,加入30ml甲醇超声(150w,44khz)30min,过滤,滤渣加入30ml甲醇超声30min,过滤,滤纸用少量甲醇潤洗,水浴蒸干滤液,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
称取0.2g赤灵芝提取物对照品用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
分别称取灵芝酸A、B、C2、G对照品5mg,用甲醇溶解定容至5ml容量瓶中。过0.22um微孔滤膜,即得。
5.4液相色谱条件
色谱柱: TSK gel ODS-80Tm(5μm 150*4.6mm)
流动相: 按下表比例,梯度洗脱
流速:1.2mL/min;柱温:25℃;检测波长:254nm进样量:10μl
5.5实验结果
用赤灵芝提取物对照品标定赤灵芝药材中灵芝酸的含量与用灵芝酸化学对照品标定赤灵芝药材中灵芝酸的含量的数据见表5。从表5数据可以看出:用两种标定方法标定赤灵芝药材中的灵芝酸含量其含量相差均小于2%,RSD值均小于1%,分离度均大于0.9,理论塔板数均大于6000。因此可以用赤灵芝提取物对照品替代灵芝酸A、灵芝酸B、灵芝酸C2、灵芝酸G四个化学对照品作为对照物检测赤灵芝中的灵芝酸含量。
表5两种不同标定方式标定灵芝酸含量的数据
实施例6用赤灵芝提取物对照品标定赤灵芝药材含量及中国药典总三萜检测方法的局限性
6.1.实验概述
用中国药典中的三萜及甾醇的检测方法测定赤灵芝中的灵芝三萜与使用本发明的赤灵芝药材的检测方法测定赤灵芝药材灵芝三萜含量进行对比,两种检测方法如下:
6.2.使用中国药典方法测定赤灵芝药材中三萜及甾醇含量
6.2.1试剂
齐墩果酸对照品购自上海源叶生物科技有限公司
6.2.2.对照品溶液及供试品溶液样品制备
取灵芝药材粉末2g,精密称定,置具塞锥形瓶中,加乙醇40ml,超声处理(功率140W,频率42kHz)30分钟,滤过,滤液置50ml量瓶中,用乙醇分次洗涤滤器和滤渣,洗液并入同一量瓶中,加乙醇至刻度,摇匀,即得。
取齐墩果酸对照品适量,精密称定,加甲醇制成每1ml含0.2mg的溶液,即得。
6.2.3.标准曲线制备
精密量取对照品溶液0.1、0.2、0.3、0.4、0.5ml,分别置15ml具塞试管中,挥干,放冷,精密加入新配制的香草醛冰醋酸溶液(精密称取香草醛0.5g,加冰醋酸使溶解成100ml,即得)0.2ml、高氯酸0.8ml,摇匀,在70℃水浴中加热15分钟,立即置冰浴中冷却5分钟,取出,精密加入乙酸乙酯4ml,摇匀,以相应试剂为空白,照紫外一可见分光光度法,在546nm波长处测定吸光度,以吸光度为纵坐标、浓度为横坐标绘制标准曲线。
6.2.4.测定法
精密量取供试品溶液0.2ml,置15ml具塞试管中,照标准曲线制备项下的方法,自“挥干”起,同法操作,测定吸光度,从标准曲线上读出供试品溶液中齐墩果酸的含量,计算,即得。
6.3.本发明的赤灵芝药材的检测方法测定赤灵芝药材灵芝三萜含量
6.3.1样品制备
称取2g药材,加入30ml甲醇超声(150w,44khz)30min,过滤,滤渣加入30ml甲醇超声30min,过滤,滤纸用少量甲醇潤洗,水浴蒸干滤液,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
6.3.2.液相色谱条件
色谱柱: TSK gel ODS-80Tm(5μm 150*4.6mm)
流动相: 按下表比例,梯度洗脱
流速:1.2mL/min;柱温:25℃;检测波长:254nm进样量:10μl
6.3.3.总灵芝三萜的计算
将赤灵芝药材样品液相色谱检测中,保留时间在15~65分钟区间的所有紫外吸收峰面积求和,并与赤灵芝提取物对照品中灵芝酸A所对应的紫外吸收峰面积相比较,算得赤灵芝药材样品中总灵芝三萜的含量。
6.4.本发明优化后的灵芝药材薄层色谱鉴别方法检测赤灵芝药材
6.4.1.样品制备方法
分别称取2g灵芝药材样品,加入30ml甲醇超声(150w,44khz)30min,过滤,滤渣加入30ml甲醇超声30min,过滤,滤纸用少量甲醇潤洗,水浴蒸干滤液,用甲醇溶解并定容至2ml容量瓶中。即得。
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。即得。
6.4.2.薄层色谱条件:
薄层板: 硅胶薄层板
点样: 点样量2μl
展开剂: 甲苯∶乙酸乙酯∶甲醇∶甲酸=15∶15∶1.0∶0.1
检视: 喷加10%硫酸乙醇试剂,烘干,在紫外灯光UV366nm下检视。
6.5.实验结果:
用中国药典中的三萜及甾醇的检测方法测定赤灵芝中的灵芝三萜与使用本发明的赤灵芝药材的检测方法测定赤灵芝药材灵芝三萜含量进行对比,含量数据表及图谱见表6及图4、图5、图6和图7。从表6可以看出:赤灵芝药材样本1-13中三萜和甾醇含量在0.8%-1%之间,各样品灵芝三萜含量差距不明显,专属性差,无法体现出不同药材质量的好坏。从图6、图7的液相色谱图和薄层色谱图中可以看到各样品间质量的明显差异,从表6可以看出:赤灵芝药材样本1-13中灵芝酸含量最高的为样本11,灵芝酸含量在0.4%以下的有6个样本,灵芝酸含量在0.4%-0.9%的有5个样本。上述结果与图6液相色谱图和图7薄层色谱图一致。因此,使用本发明的赤灵芝提取物对照品为对照测定赤灵芝药材灵芝三萜含量比中国药典所述方法专属性强,能够显示赤灵芝药材的质量差异,更精确判别出赤灵芝药材质量的好坏。
表6:使用2015版中国药典方法检测赤灵芝药材三萜及甾醇含量及使用本发明的赤灵芝药材的检测方法测定赤灵芝药材灵芝三萜含量
图6中,图谱信号由下至上分别是如下样品:赤灵芝药材1、赤灵芝药材2、赤灵芝药材3、赤灵芝药材4、赤灵芝药材5、赤灵芝药材6、赤灵芝药材7、赤灵芝药材8、赤灵芝药材9、赤灵芝药材10、赤灵芝药材11、赤灵芝药材12、赤灵芝药材13;
图7中,谱带标号由左至右分别为:1、赤灵芝提取物对照品;2、赤灵芝药材1;3、赤灵芝药材2;4、赤灵芝药材3;5、赤灵芝药材4;6、赤灵芝药材5;7、赤灵芝药材6;8、赤灵芝药材7;9、赤灵芝药材8;10、赤灵芝药材9;11、赤灵芝药材10;12、赤灵芝药材11;13、赤灵芝药材12;14、赤灵芝药材13。
图4、图5中的横坐标数字对应表6中的药材编号。
实施例7用赤灵芝提取物对照品测定赤灵芝药材灵芝三萜含量并对赤灵芝药材进行质量评级
7.1实验概述
用赤灵芝提取物对照品及液相色谱法测定赤灵芝药材中灵芝三萜含量,方法如下:
7.2样品制备
称取2g药材,加入30ml甲醇超声(150w,44khz)30min,过滤,滤渣加入30ml甲醇超声30min,过滤,滤纸用少量甲醇潤洗,水浴蒸干滤液,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
称取0.2g赤灵芝提取物对照品,用甲醇溶解并定容至2ml容量瓶中。样品过0.22um微孔滤膜,即得。
7.3液相色谱条件
色谱柱: TSK gel ODS-80Tm(5μm 150*4.6mm)
流动相: 按下表比例,梯度洗脱
流速:1.2mL/min;柱温:25℃;检测波长:254nm进样量:10μl
7.4总灵芝三萜的计算
将赤灵芝药材样品液相色谱检测中,保留时间在15~65分钟区间的所有紫外吸收峰面积求和,并与赤灵芝提取物对照品中灵芝酸A所对应的紫外吸收峰面积相比较,算得赤灵芝药材样品中总灵芝三萜的含量。
7.5实验结果
用赤灵芝提取物对照品测定赤灵芝药材中灵芝三萜含量的数据见表7及图8。从表7可以看出:测定的13个样本中赤灵芝药材中编号为3号和11号的样本灵芝三萜含量远高于其他样本,含量超过13mg/g,根据质量标准定为特级品;编号为10号的样本的灵芝三萜含量在8mg/g-13mg/g之间,定为一级品;编号为9、12、13号的样本灵芝三萜含量在5mg/g-8mg/g,定为二级品;编号为1、2、4、5、6、7、8号的样本灵芝三萜含量在1mg/g-5mg/g,定为三级品。因此可以明显看出:使用本发明的方法可以精细的判别出赤灵芝药材质量的优劣,并且可以很好的对各地质量差异很大的灵芝药材进行分类评级,从而保证了灵芝药材的质量。
表7:用赤灵芝提取物对照品测定赤灵芝药材中灵芝三萜含量
注:药材分级标准:灵芝三萜含量大于13mg/g,该药材为特级品
灵芝三萜含量在8-13mg/g范围,该药材为一级品;
灵芝三萜含量在5-8mg/g范围,该药材为二级品;
灵芝三萜含量在小于1-5mg/g范围,该药材为三级品;
灵芝三萜含量在小于1mg/g,该药材为等外品;
以上所述,仅是本发明的较佳实施例而已,并非对本发明做任何形式上的限制,故凡未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!