定量分析方法与流程
[相关申请的交叉引用]
本申请要求于2016年5月2日提交到韩国知识产权局的韩国专利申请no.10-2016-0054142的权益,该申请的公开内容通过引用全部并入本文。
本发明涉及一种使用nmr光谱定量分析样品的方法。
背景技术:
可以通过多种方法对可溶性样品进行定量分析。作为一个实例,在进行色谱分离之后,可以基于光谱检测(例如,uv/vis、elsd等)或频谱检测(例如质谱仪)来分析样品。nmr最常用于化学结构的分析,但是逐渐认为,其对于定量分析也十分重要。由于信号的积分强度与信号上表现出的核的数量成正比,因此nmr可以用于定量分析。此外,由于光谱中所有质子的敏感度相同,因此具有定量分析不需要消光系数或验证/校准的优点。
在使用nmr的定量分析中最常用的方法是使用内标物的方法。换句话说,是如下方法:将已知分子结构和用量的标准物质与样品放在一起,比较并定量分析各个信号的积分强度。这种方法具有定量分析简单且精确的优点,但是具有不能应用于不溶性样品的缺点。
对于不溶性样品,可以应用ssnmr(固态nmr),但是使用内标物的定量分析具有局限性。其原因如下:1)内标物和样品应当均匀混合,但是在不溶性样品的情况下,难以均匀混合;2)在混合的过程中,标准物质或样品残留在混合工具中,因此,难以进行精确的定量分析;3)由于不均匀混合而产生旋转误差;以及4)nmr峰呈现较宽峰,因此,难以选择可与样品区分开的内标物。
近来,已经积极地对无机-无机复合物质以及无机物质进行了研究。然而,由于这些物质大多数是不溶性物质,因此需要它们的定量方法。然而,在这种不溶性物质的常规定量分析中,已经使用诸如tga等方法,但是具有可靠性降低、定量分析方法复杂以及需要大量时间的问题。
因此,本发明人对样品的定量分析方法,特别是不溶性样品的定量分析方法进行了深入研究。结果,本发明人发现,如下面将描述的,通过使用获得样品和标准物质的nmr光谱然后对其进行比较的外标法进行定量分析,其中,可以从各自的nmr光谱的信息来定量分析样品。基于这样的发现而完成本发明。
技术实现要素:
技术问题
本发明的目的是提供一种使用nmr光谱定量分析样品的方法。
技术方案
为了实现上述目的,本公开提供一种样品中的化合物的定量方法,包括以下步骤:
1)获得含有化合物中所含的nmr活性原子的标准物质的nmr活性原子的nmr光谱,并在相同条件下获得该化合物的nmr活性原子的nmr光谱;
2)分别得到标准物质的nmr光谱和样品的nmr光谱中的特征峰的fid(自由感应衰减)振幅值;以及
3)比较各个fid振幅值以测定样品中的所述化合物的浓度。
本发明用于样品中包含的特定化合物的定量分析,其特征在于使用nmr光谱进行定量。除非本文另外说明,否则样品中的化合物是指作为样品中包含的化合物待进行定量分析的化合物。特别地,作为样品和其中包含的化合物,不仅可以使用可溶性物质,而且可以使用不溶性物质。
通常,由于nmr光谱中的信号的积分强度与信号上表现出的核素成比例,因此可以基于待定量化合物的特定信号进行定量。换句话说,当将分子结构已知的标准物质与样品放在一起用于精确定量分析时,在nmr光谱中可以得到标准物质的信号的积分强度和待定量化合物的特定信号的积分强度。在这种情况下,由于标准物质的分子结构和用量已知,因而可以定量分析样品中的化合物。因此,将与样品放在一起的标准物质称为“内标物”。
然而,上述定量分析具有难以应用于不溶性样品的问题。最主要的原因是,为了得到有效的nmr光谱,样品和标准物质应该均匀混合,但是对于不溶性样品,其在溶剂中不混合,因此难以混合均匀。
在这一方面,本发明使用分别得到标准物质和样品的nmr光谱然后分析它们的方法,而不是如上所述将标准物质与样品混合,将本文所使用的标准物质称为“外标物”。
下文中,将详细描述本发明的各个步骤。
本发明的步骤1是分别得到标准物质和样品的nmr光谱的步骤。
由于分别得到nmr光谱并对其进行比较,因此样品中所含的化合物和标准物质必须含有相同的nmr活性原子。nmr活性原子可以是,例如,氢、锂、碳、氟、硅、磷、铅或锡。
基于nmr活性原子得到样品和标准物质的nmr光谱。例如,当使用13c作为参比时,分别得到样品和标准物质的13cnmr。另外,本发明中涉及的nmr光谱包括ssnmr(固态nmr)。例如,当nmr活性原子设定为207pb时,分别得到样品和标准物质的207pbssnmr。而且,由于通过得到样品和标准物质的nmr光谱并对其进行比较来进行定量分析,因此得到nmr光谱的条件应该相同。上述条件是指nmr测定所需的条件,例如,是指当进行nmr测定时,扫描次数、延迟时间、脉冲宽度、脉冲功率、接收器增益和旋转速率相同。此外,对每个条件的范围没有限制,只要它适合于在相同的条件下分别得到nmr光谱即可。
另一方面,对于标准物质,由于可以知道用于得到nmr光谱的标准物质的分子结构、分子量和质量,因此可以在后面利用它们进行定量分析。此外,对于标准物质,对其类型没有特别地限制,只要在nmr光谱中出现特征峰即可,优选地,使用在nmr光谱中特征峰显著呈现的物质。例如,当获得13cnmr光谱时,标准物质优选为hmb(六甲基苯),当获得207pbnmr光谱时,标准物质优选为pb(no3)2。
本发明的步骤2是获得在上述步骤1中得到的各个nmr光谱中的特征峰的fid振幅值的步骤。
nmr光谱以磁场中观察到的电信号即自由感应衰减(fid)为基础。由于fid本身难以分析,因此将其转换为光谱。在这一过程中,进行相位调整、基线校正和峰积分。在nmr光谱中,特定峰的积分强度与出现在该峰处的核的数量成正比,因此可以用于定量分析。然而,当在nmr光谱中许多峰重叠时,难以获得积分强度,因此难以定量分析。
然而,相反地,可以在nmr光谱的特定峰区域提取fid。在这种情况下,可以提取fid振幅值。这些fid振幅值包含nmr光谱中的特定峰上的定量信息,因此通过提取fid振幅值可以进行定量分析。例如,可以使用agilent的vnmrj4.2软件得到fid振幅值。
也就是说,标准物质的nmr光谱的特定峰区域中的fid振幅值与在该峰处表现出的核素的量成正比,在与标准物质的nmr光谱相同的条件下得到样品的nmr光谱,可以将上述比例关系应用至样品的fid振幅值。因此,如果基于待定量化合物的特定峰从样品的nmr光谱中得到fid振幅值,通过将其与标准物质的fid振幅值进行比较可以定量分析所述化合物。
本发明的步骤3是根据上述步骤2中得到的各个fid振幅值分析样品中的化合物的浓度的步骤。
在上述步骤2中,如果从标准物质的nmr光谱得到特征峰的fid振幅值,标准物质的分子结构以及其用量已知,因此可以基于其fid振幅值进行定量分析。
具体而言,各个fid振幅值与标准物质或样品中化合物的nmr活性原子的数量成比例。因此,如下面的式(1)所示,通过比较fid振幅值可以测定样品中的化合物的浓度。
[式1]
样品中的化合物的浓度(重量%)=(a/b)×(c/d)×(e/f)×(g/h)
在式1中,
a是对应于标准物质的特征峰的标准物质分子中nmr活性原子的数量,
b是对应于样品的特征峰的化合物分子中nmr活性原子的数量,
c是化合物的分子量,
d是标准物质的分子量,
e是用于获得标准物质的nmr光谱的标准物质的质量,
f是用于获得样品的nmr光谱的样品的质量,
g是样品的fid振幅值,
h是标准物质的fid振幅值。
在式1中,a至f是在获得nmr光谱之前可以得到的值,g和h是可以通过上述步骤1和2得到的值。
作为一个实例,在下面描述的实施例1的情况下,使用hmb(六甲基苯)作为标准物质,使用hmb和adm(金刚烷)的混合物作为样品,在相同的条件下获得13cnmr光谱,然后通过提取每个甲基峰的fid振幅值来定量分析hmb。结果,几乎与实际值相等的定量分析是可能的。
另外,在下面描述的实施例2的情况下,使用pb(no3)2作为标准物质,使用pbi2(dmso)和pbi2的混合物作为样品,在相同的条件下获得207pbnmr光谱,然后分别提取pb峰的fid振幅值并定量分析。结果,几乎与通过tga确定的值相等的定量分析是可能的。
如上所述,在本发明中,在通过使用外标物的方法得到nmr光谱之后,可以从光谱上的信息定量地分析样品中的化合物,并且也可以应用于可溶性样品以及不溶性样品。
有益效果
本发明提供一种样品中的化合物的定量分析方法,其特征在于,通过获得样品和标准物质的nmr光谱然后对它们进行比较的使用外标物的方法进行定量分析,所述方法甚至可以应用于不溶性样品。
附图说明
图1示出了本发明的实施例1中的hmb50样品的fid振幅值的提取结果;
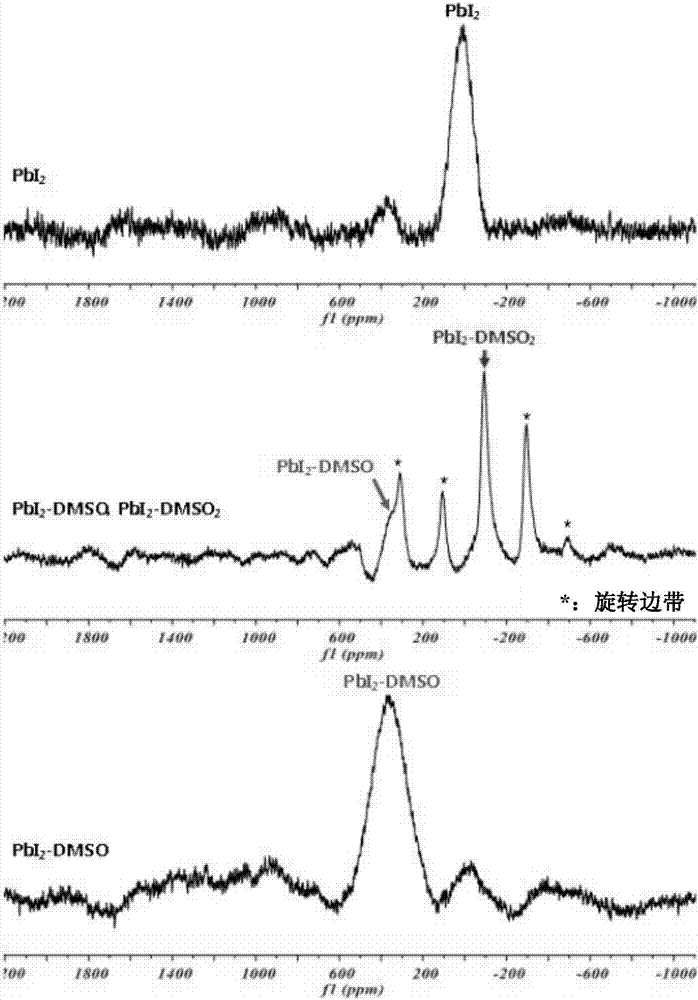
图2示出了本发明的实施例2中的pbi2、第一样品和第二样品的207pbssnmr光谱;
图3示出了本发明的实施例2中的pb(no3)2的207pbssnmr光谱;
图4示出了本发明的实施例2中的pbi2(dmso)2的特征峰的fid振幅值的提取结果;
图5示出了本发明的实施例2中的pbi2(dmso)的特征峰的fid振幅值的提取结果;
图6示出了本发明的实施例2中的第二样品的tga结果。
具体实施方式
下文中,将提供优选实施例以帮助理解本发明。然而,提供下面的实施例仅用于说明的目的,本发明的内容不局限于这些实施例。
在下面的实施例中,除非另外说明,否则nmr光谱使用agilentdd2600mhzssnmr(使用1.6mm的ssnmr探针),并且使用agilent的vnmrj4.2软件得到fid振幅值。
实施例1:样品中的hmb的定量
为了验证根据本发明的定量方法,使用hmb(六甲基苯)和adm(金刚烷)进行实验。使用hmb(100重量%的hmb)与外标物分别制备样品hmb50(adm:hmb=50:50(重量%))和样品hmb30(adm:hmb=70:30(重量%))。在nmr转子中采样的外标物为19.48mg,hmb50和hmb30分别为19.89mg和19.88mg。
在下面的条件下分别得到它们的nmr光谱,并且分别提取对应于hmb的甲基的特征峰的fid振幅值。此时,将延迟时间分别变为1秒、5秒和30秒,并进行实验。
-脉冲宽度=90度脉冲
-扫描次数=16
-接收器增益=24
-旋转速率=10khz
hmb50的fid振幅值的提取结果的一个实例示于图1中,剩余样品的fid振幅值的提取结果及其定量结果示于下面的表1中。
[表1]
在上面的表1中,使用上面描述的式1计算定量值。例如,在延迟时间=30秒的hmb50的情况下,当代入上面描述的式1时,定量值如下。
定量值(重量%)=(a/b)×(c/d)×(e/f)×(g/h)
a(hmb分子中的甲基数)=6
b(hmb分子中的甲基数)=6
c(hmb分子量)=162.28g/mol
d(hmb分子量)=162.28g/mol
e(nmr转子中采样的外标物的质量)=19.48mg
f(nmr转子中采样的hmb50的质量)=19.89mg
g(外标物的fid振幅值)=18.5
h(hmb50的fid振幅值)=9.6
如上所述,可以证实,充足的延迟时间改善了定量性,并且误差不大。
实施例2:样品中的pbi2(dmso)2的定量
1)样品的制备
已经报道了使用dmso加合物(pbi2(dmso)2)作为钙钛矿前体来制备用作太阳能电池的光吸收剂的钙钛矿的一个实例(science2015,第348卷,第6240期,第1234至1237页)。根据该文献,将pbi2溶解在dmso中以生成中间体,然后加热以制备pbi2(dmso)。然而,上述中间体混合在(pbi2(dmso)2)和pbi2(dmso)中,并且它们各自的含量范围根据制备条件而变化。为了制备高纯度的pbi2(dmso),必须确认中间体中的pbi2(dmso)2的浓度。
因此,根据上述文献,在60℃下将pbi2(50g)溶解在150ml的dmso中,然后滴加350ml的甲苯。随后,将沉淀物过滤并干燥3小时,取其一部分作为“第一样品”。将除第一样品之外的剩余样品置于60℃的真空烘箱中24小时以制备pbi2(dmso),取其一部分作为“第二样品”。
2)特征峰的选择
为了在第一样品和第二样品中选择pbi2(dmso)2和pbi2(dmso)的特征峰,在agilentdd2600mhz中使用3.2mm的ssnmr探针,在下面的条件下得到在上述制备中使用的pbi2、第一样品和第二样品的207pbssnmr光谱,结果示于图2中。
-脉冲功率(tpwr)=55
-脉冲宽度(pw)=5.00微秒
-ax90=3500
-延迟时间=5秒
-扫描次数=50000
-接收器增益=60
-旋转速率=25khz
如图2中所示,pbi2(dmso)2和pbi2(dmso)存在于第一样品中,并且取pbi2(dmso)2的峰作为特征峰。此外,pbi2(dmso)2和pbi2(dmso)存在于第二样品中,并且取pbi2(dmso)的峰作为特征峰。
3)标准物质的fid振幅值的提取
然后,使用pb(no3)2(100重量%的pb(no3)2,32mg)作为207pb标准物(外标物)。使用agilentdd2600mhzssnmr(使用1.6mm的ssnmr探针)在下面的条件下得到207pbssnmr光谱。从pb(no3)2的nmr光谱提取pb的特征峰的fid振幅值,结果示于图3中。
-脉冲功率(tpwr)=60
-脉冲宽度(pw)=90度脉冲(2.25微秒)
-ax90=3500
-延迟时间=5秒
-扫描次数=5000
-接收器增益=60
-旋转速率=35khz
4)第一样品中的pbi2(dmso)2的定量
使用21.98mg的第一样品,在与上述3)中的条件相同的条件下得到207pbssnmr光谱。在上述光谱中,提取之前确认的pbi2(dmso)2的特征峰的fid振幅值,结果示于图4中。
使用上述结果,如下面的表2中所示定量第一样品中的pbi2(dmso)2。
[表2]
5)第二样品中的pbi2(dmso)2的定量
使用21.36mg的第二样品,在与上述3)中的条件相同的条件下得到207pbssnmr光谱。在上述光谱中,提取之前确认的pbi2(dmso)的特征峰的fid振幅值,结果示于图5中。
使用上述结果,如下面的表3中所示定量第二样品中的pbi2(dmso)。
[表3]
6)定量分析的验证
为了验证上述定量分析的结果,通过在science2015,第348卷,第6240期,第1234至1237页中描述的tga方法分析浓度。
tga方法是对dmso的量进行定量的方法,当如在第二样品中存在pbi2(dmso)和pbi2时,可以进行定量分析,但是当如在第一样品中存在pbi2(dmso)和pbi2(dmso)2时,无法确定检测到dmso是由哪种结构产生的。因此,仅第二样品通过tga方法定量,结果示于图6中。
第二样品的tga结果示于图6中。由于pbi2在600℃以上分解,因此可认为至300℃时的重量减少是由于dmso。tga的起始质量为7.401mg,至300℃时的重量减少为1.038mg。因此,第二样品中的dmso的质量为1.038mg,换算成摩尔数(dmso的分子量:78.13)为0.000013286摩尔。由于第二样品中的dmso存在于pbi2(dmso)中,因此上述摩尔数与pbi2(dmso)的摩尔数相等,因此换算成质量为7.163mg,并且第二样品中的pbi2(dmso)为96.78重量%(7.163mg/7.401mg)。
可以证实,上述定量结果与根据本发明的定量方法分析得到的第二样品中的pbi2(dmso)的浓度96.6重量%非常相近。
- 还没有人留言评论。精彩留言会获得点赞!