一种鉴别味连、雅连、凤尾连的方法与流程
本发明中药鉴别领域,特别是涉及一种黄连的鉴别方法。
背景技术:
黄连(coptidisrhizoma)是一味常用中药,疗效确切,临床上应用广泛,始载于东汉《神农本草经》,列为上品,据不完全统计,我国古代方术记载的3万多中医处方中,含有黄连的处方就有1760个,约5%左右的方剂中有黄连,据《全国中成药品种目录》统计,以黄连为原料的中成药品种有黄连上清丸、复方黄连素片、加味香连丸、香连丸等100多种[i]。《中国药典》(2015版)收载黄连为毛茛科植物黄连coptischinensisfranch、三角叶黄连coptisdeltoideac.y.chengethsiao.或云连coptisteetawall.的干燥根茎。系秋季采挖,除去须根及泥沙,干燥,撞去残留须根。黄连因基源不同,药材分为“味连”、“雅连”和“云连”。产自四川的黄连主要为“味连”和“雅连”,除此之外,峨眉野连coptisomeiensis(chen)c.y.cheng.也为四川地区的历史习用品种,习称“凤尾连”。
由于黄连的品种较多,在生产、实践中由于一些经济、药效等原因经常需要确认其品种,中药鉴别常用的方法有光谱鉴别法,色谱鉴别法,显微镜鉴别法等。
光谱与分子结构密切相关,是研究和表征分子结构的一种手段,常用于物质的鉴别。但大分子或混合物体系的谱图是比较复杂的,谱峰难以指认,解谱困难。
色谱法此法具有快速、经济、可靠、操作简单、适用范围广、重现性好等优点,为国内外学者最快接受和广为应用是理所当然的。在中药新药研制中,几乎所有的新药都必须提供薄层层析谱,并需附有标准品或阴阳药材对照的彩色照片。但在实际的薄层层析鉴别中,因中药的成分性质相近而难以分开。
显微鉴别是利用显微镜来观察药材的组织构造,细胞形状以及内含物的特征,用以鉴定药材真伪、纯度甚至品质。通常应用于单凭性状不易识别的药材、破碎药材、粉末药材以及丸、散、膏、丹等中药成方制剂。显微观察主要包括横切片或纵切片观察、表面片观察、粉末观察、解离组织观察、显微测定、偏振光显微镜观察等几个方面。显微鉴别的主要工具为显微镜,只有熟悉显微镜的相关知识,才能正确使用显微镜,从而发挥仪器的性能,手工绘图需要技巧,否则便易失真,显微描绘器又易受视场限制,而影响图画的位置和清晰度。
综上所述,现有的方法存在一定的局限性,不能满足鉴别黄连的品种的要求,本发明研究了一种新的方法鉴别黄连的品种。
技术实现要素:
本发明拟提供一种可以用于鉴别雅连、味连和凤尾连的方法。
经研究发现,雅连、味连和凤尾连中含量最高的都是小檗碱,且7种生物碱分别的含量大小具有以下规律。
味连:小檗碱>黄连碱>巴马汀>表小檗碱>药根碱>非洲防己碱>格兰地新(groenlandicine),且不同批次间之间差异较小;
雅连:小檗碱>黄连碱>巴马汀>药根碱>格兰地新(groenlandicine)>表小檗碱>非洲防己碱;
凤尾连:小檗碱>黄连碱>药根碱>巴马汀>格兰地新(groenlandicine)>非洲防己碱>表小檗碱。
3种川产黄连所含7种生物碱比例各不相同,据此可根据7种生物碱含量比例差别,来鉴别3种品种。
基于上述发现,本发明实际提供了一种可以将味连/雅连、味连/凤尾连、雅连/凤尾连区分鉴别的方法,其步骤主要包括:取待检药材,测定小檗碱、非洲防己碱、格兰地新groenlandicine、表小檗碱、药根碱、黄连碱、巴马汀的含量,将上述各成分的含量大小进行比较,即可得出鉴别结果:
当药材中成分含量为小檗碱>黄连碱>巴马汀>表小檗碱>药根碱>非洲防己碱>格兰地新时,该药材为味连;
当药材中成分含量为小檗碱>黄连碱>巴马汀>药根碱>格兰地新>表小檗碱>非洲防己碱时,该药材为雅连;
当药材中成分含量为小檗碱>黄连碱>药根碱>巴马汀>格兰地新>非洲防己碱>表小檗碱时,该药材为凤尾连。
本发明中还发现,雅连中的格兰地新含量高于味连或凤尾连。
本发明中可以采用高效液相色谱法进行含量测定,其中,流动相选自乙腈-含有缓冲盐的水相(30~34:66~77);色谱柱填料选自十八烷基键合硅胶。
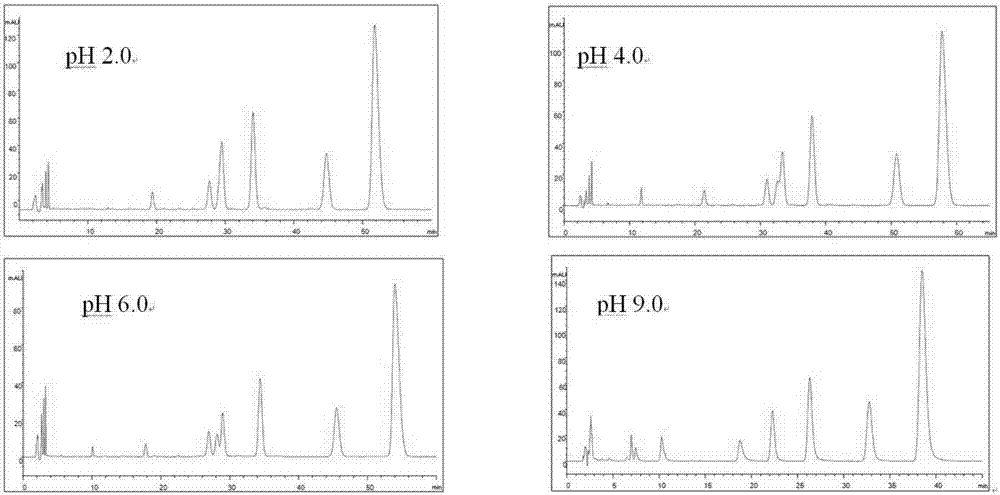
进一步地,高效液相色谱法中含有缓冲盐的水相ph为6.0~9.0。实验表明,在ph2.0时,表小檗碱和药根碱峰完全重合;在ph4.0时,表小檗碱和药根碱略有分开;在ph6.0时,表小檗碱和药根碱的分离度分别小于1.5。在ph9.0时,表小檗碱和药根碱的分离度分别大于1.5。
本发明中,所述缓冲盐可以选自醋酸盐、碳酸盐、磷酸盐。
具体实施方式中,所述醋酸盐选自醋酸铵,所述碳酸盐选自碳酸氢钠,所述磷酸盐选自磷酸二氢钾或磷酸二氢钠。
实验表明,以醋酸铵或碳酸氢铵为缓冲盐时,格兰地新(groenlandicine)、表小檗碱、非洲防己碱、药根碱等峰均出现特别严重的拖尾和变形,峰形不佳,而以磷酸二氢钾为缓冲盐的流动相,格兰地新(groenlandicine)、表小檗碱、非洲防己碱各峰稍有拖尾,峰形较佳,因此选择磷酸二氢钾为缓冲盐的流动相。
本发明的一个具体实施方式中,所述含有缓冲盐的水相为,乙腈-0.05mmol/l磷酸二氢钾水溶液(30~34):(66~77),其中,磷酸二氢钾水溶液中还含有0.4%十二烷基磺酸钠。
本发明的一个具体实施方式中,色谱柱选自angilenteclipsexdb-c18,250mm×4.6mm,5μm。
本发明中,检测波长为270nm或345nm,或其附近波长。但是,在允许的误差范围内的波长均应纳入本发明的保护范围,而这一允许的误差范围可以通过简单的替换实验获取。如343、344、346、347nm等,当然不限于本发明的举例。
进一步,所述高效液相色谱法的流动相为,乙腈-0.05mmol/l磷酸二氢钾(含有0.4%十二烷基磺酸钠,ph9.0)(30~34:66~77)。
当采用流动相比例30:70,分析一个待测样品时间为75min;采用流动相比例32:68,分析一个待测样品时间为55min;采用流动相比例(34:66),分析一个待测样品时间为45min,在保证分离度和峰形的基础上选择流动相比例为34:66,可以节省时间,同时能够减少流动相使用量和仪器损耗,从而降低检测成本。
另外,高效液相色谱中的柱温、进样量和流速,均可按照正常范围内取值,当然不同值对检测结果会有细微影响,具体使用时可以根据具体情况进行常规操作以便确定实际使用条件。本发明一个具体实施方式中,柱温35℃,检测波长345nm,进样量10μl。
当然,上述方法是通过测定7种成分含量来进行鉴别区分,经过发明人进一步地分析发现,小檗碱和黄连碱两者在三种植物中的成分排序几乎是恒定的,也是相同的,不会对三种植物的鉴别带来影响,而其余5种成分却在三种植物中有差异,因此,为了能够进一步节省检测成本,本发明提供了一种成本更低廉、步骤更简便的鉴别方法,包括如下操作:
取待检药材,测定非洲防己碱、格兰地新groenlandicine、表小檗碱、药根碱、巴马汀的含量,将上述各成分的含量大小进行比较,即可得出鉴别结果:
当药材中成分含量为巴马汀>表小檗碱>药根碱>非洲防己碱>格兰地新时,该药材为味连;
当药材中成分含量为巴马汀>药根碱>格兰地新>表小檗碱>非洲防己碱时,该药材为雅连;
当药材中成分含量为药根碱>巴马汀>格兰地新>非洲防己碱>表小檗碱时,该药材为凤尾连。
经方法学验证,采用本发明高效液相色谱法的色谱条件进行含量测定试验线性范围大、精密度佳、重现性好、准确、稳定。
本明的有益效果是:不同种类的黄连其成份数量,种类,性质相近,用现有的方法难以区分,使用本发明的方法可以高效的鉴别黄连的品种。
1)快速准确,用高效液相法可以快速的测定黄连成份的含量,使用本发明的鉴别方法分离效率高,选择性好,检测灵敏度高,操作自动化,使得黄连成份含量准确度高,进而准确鉴别其品种。
2)简便,本发明方法采用黄连所含生物碱的量各不相同,来鉴别品种,可以形成各黄连品种生物碱数据库,使鉴别过程更加简便。
附图说明
图1是7种黄连生物碱的紫外吸收图;
图2是不同ph选择的色谱图;
图3是不同缓冲盐选择的色谱图;
图4是不同流动相比例色谱图;
图5是a.混合对照品,b.味连,c雅连;
图6是6批味连药材生物碱含量测定色谱叠加图;
图7是6批雅连药材生物碱含量测定色谱叠加图;
图8是3批凤尾连药材生物碱含量测定色谱叠加。
具体实施方式
实施例1
1.1仪器与试剂
angilent1260-ii型高效液相色谱仪;bs-110s电子分析天平(北京赛多利斯电子天平有限公司);超声波清洁器(昆山市超声仪器有限公司)。
groenlandicine、表小檗碱、药根碱、黄连碱、巴马汀购于成都普思生物生物科技有限公司(批号分别为ps161130-03、ps161130-01、ps161124-01、ps161201-01、ps161130-02,含量≥98%),非洲防己碱购于成都曼斯特生物科技有限公司(批号为must-14072213,含量≥98%),盐酸小檗碱购于中国食品药品检定研究院(批号为110713-200911,含量≥98%)。
甲醇、乙腈(色谱纯,sigma);三乙胺、磷酸、十二烷基苯磺酸钠、磷酸二氢钾均为分析纯。
1.2实验药材
本课题组野外采集黄连药材15批,含味连6批,雅连6批,凤尾连3批,均由西南交通大学生命科学与工程学院宋良科教授鉴定为黄连coptischinensis(味连)、三角叶黄连coptisdeltoidea(雅连)、峨眉野连coplisomeiensis(凤尾连)。详细产地及编号见表1-1。
表1-1黄连产地及编号
1.3实验方法
1.3.1对照品溶液的制备
分别精密称定7种对照品适量,加盐酸甲醇溶液(1:100),溶解制得每1ml含有1mg的对照品储备液。精密吸取各对照品储备液适量,配制成每1ml分别含格兰地新(groenlandicine)、表小檗碱、非洲防己碱、药根碱、黄连碱、巴马汀、小檗碱、为0.026、0.043、0.030、0.031、0.019、0.029、0.067mg/ml的混合对照品溶液。
1.3.2供试品溶液的制备
取黄连粉末约0.1g(过三号筛),精密称定,置具塞锥形瓶中,精密加入25ml盐酸甲醇(1:100),密塞,称定重量,超声提取45min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,放置,取上清液,过0.45μm微孔滤膜,即得供试品溶液。
1.3.3色谱条件
1.3.3.1波长选择
小檗碱(berberine)、黄连碱(coptisine)、表小檗碱(epiberberine)、巴马汀(palmatine)、药根碱(jatrorrhizine)、非洲防己碱(columbamine)、格兰地新(groenlandicine)紫外吸收图谱见图1。
由图1可见,7种黄连生物碱的紫外吸收在270、345nm附近出较为接近,故根据文献选择345nm作为检测波长。
1.3.3.2流动相ph的选择
以乙腈-0.4%的磷酸二氢钾(含0.4%十二烷基苯磺酸钠,分别用磷酸、三乙胺调节ph2.03.0、4.0、6.0、9.0)34:66为流动相,流速:1ml/min,柱温:35℃,检测波长:345nm,分别进样,测得的色谱图如图2。
由图2可见,在ph2.0时,表小檗碱和药根碱峰完全重合;在ph4.0时,表小檗碱和药根碱略有分开;在ph6.0时,表小檗碱和药根碱的分离度分别为0.94和1.34。在ph9.0时,表小檗碱和药根碱的分离度分别为11.59、4.63。故选择磷酸二氢钾调节ph9.0。
1.3.3.3缓冲盐的选择
分别以乙腈-0.05mmol/l磷酸二氢钾(含有0.4%十二烷基磺酸钠,ph9.0)(34:66)、乙腈-0.05mmol/l醋酸铵(含有0.4%十二烷基磺酸钠,ph9.0)(34:66)、乙腈-0.05mmol/l碳酸氢铵(含有0.4%十二烷基磺酸钠,ph9.0)(34:66)为流动相,流速:1.0ml/min,柱温:35℃,检测波长:345nm,分别进样,测得的色谱图如图3。
由图3可见,以醋酸铵和碳酸氢铵为流动相groenlandicine、表小檗碱、非洲防己碱、药根碱等峰均出现特别严重的拖尾和变形,峰形不佳,而以磷酸二氢钾为流动相,groenlandicine、表小檗碱、非洲防己碱各峰稍有拖尾,峰形较佳,因此选择磷酸盐缓冲液为流动相。
1.3.3.4流动相比例的选择
分别以乙腈-0.05mmol/l磷酸二氢钾(含有0.4%十二烷基磺酸钠,ph9.0)(34:66)、(32:68)、(30:70)为流动相,流速:1.0ml/min,柱温:35℃,检测波长:345nm,分别进样,测得的色谱图如图4。
由图4可见,流动相比例30:70,整个分析周期时间为75min;流动相比例32:68,整个分析周期时间为55min;流动相比例(34:66),整个分析周期时间为45min。因此选择流动相比例为34:66。
1.3.3.5色谱条件
色谱柱angilenteclipsexdb-c18(250mm×4.6mm,5μm),流动相为乙腈-0.4%磷酸二氢钾(含有0.4%十二烷基磺酸钠,0.6%三乙胺,磷酸调节ph9.0)(34:66),流速1.0ml·min-1,柱温35℃,检测波长345nm,进样量10μl。
1.3.4系统适用性试验
分别取黄连生物碱混合对照品溶液、味连、雅连供试品溶液各10μl,按1.3.3.5项下色谱条件,进样测定,记录色谱图,见图5。本试验条件下,供试品溶液色谱中与各对照品对应的吸收峰的理论塔板数均不低于4000,对应的色谱峰的分离度均大于1.5。
实验结果
精密称取过二号筛的黄连粉末0.1g,按1.3.2项方法制备供试品溶液,将供试品溶液和对照品溶液各10μl进样分析,分别测定3批不同品种黄连的生物碱含量,结果见表1-2,6批味连(s1~s6,s0为黄连生物碱混合对照品)的含量测定色谱图如图6所示;6批雅连(s7~s12,s0为黄连生物碱混合对照品)的含量测定色谱图如图7所示;3批凤尾连(s13~s15,s0为黄连生物碱混合对照品)的含量测定色谱图如图8所示。
表1-2黄连生物碱含量测定结果(%,n=3)
实施例2方法学考察
1.3.5.1线性关系考察
分别精密吸取1.3.1项下各对照品储备溶液,用盐酸甲醇(1:100)溶液分别稀释成一系列浓度的混合对照品溶液。groenlandicine浓度分别为5.273、13.18、26.37、52.73、105.46μg/ml;表小檗碱浓度分别为3.845、9.612、19.22、38.45、76.90μg/ml;非洲防己碱浓度分别为5.138、12.85、25.69、51.38、102.76μg/ml;药根碱浓度分别为8.576、21.44、42.88、85.76、171.526μg/ml;黄连碱浓度分别为3.84、9.60、19.20、38.41、76.82μg/ml;巴马汀浓度分别为12.74、31.84、63.69、127.37、254.75μg/ml;小檗碱浓度分别为13.42、33.55、67.10、134.20、268.40μg/ml。上述对照品溶液,按1.3.3.5色谱条件进样,记录峰面积,以峰面积y为纵坐标,进样量(μg)为横坐标,回归分析,得回归方程、相关系数(r)及线性范围,见表1-3。结果表明,7个生物碱成分在一定范围内呈良好的线性关系。
表1-37种成分线性关系
1.3.5.2精密度试验
精密吸取同一混合生物碱对照品溶液,按1.3.3.5项色谱条件重复进样6次,记录峰面积值,计算rsd。各生物碱峰面积rsd分别为0.97%、0.63%、0.84%、0.56%、0.54%、0.76%,表明方法有较好的精密度。结果见表1-4。
表1-4精密度考察结果(mau)
1.3.5.3重复性试验
精密称取0.1g的雅连样品6份,按1.3.2项下方法制备供试品溶液,1.3.3.5项下色谱条件分别进样,记录峰面积,计算含量,计算rsd值。所测含量的rsd分别为1.74%、1.58%、1.90%、2.16%、2.01%、2.17%、1.64%,说明本方法具有可重复操作性。结果见表1-5。表1-5重复性考察(%)
1.3.5.4稳定性试验
精密称取雅连样品0.1g,按1.3.2项下方法制备供试品溶液,分别于0、2、4、8、12、24h时间按1.3.3.5项下色谱条件进样,记录峰面积,计算rsd值。峰面积的rsd分别为1.11%、1.17%、2.89%、0.60%、0.33%、1.40%、0.63%,说明供试品溶液在24h内稳定可用。结果见表1-6。
表1-6稳定性考察结果(mau)
1.3.5.5加样回收试验
取雅连样品6份,精密称定,每份0.1g,分别精密加入等量的混合对照品溶液,按1.3.2项方法制备供试品溶液,按1.3.3.5项色谱条件进样,计算加样回收率。
格兰地新(groenlandicine)、药根碱、非洲防己碱、表小檗碱、黄连碱、巴马汀、小檗碱平均回收率分别为97.07%、99.26%、101.60、99.43、98.62%、98.67%、98.68%,rsd分别为1.07%、0.66%、0.47%、0.47%、0.27%、0.37%、0.29%。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!