一种同时测定酸式和内酯式洛伐他汀的方法与流程
本发明属于洛伐他汀的测定技术领域,具体涉及一种同时测定酸式和内酯式洛伐他汀含量的方法。
背景技术
洛伐他汀是曲霉类在生长后期产生的、能有效降低血浆胆固醇的次级代谢产物。1979年日本学者远藤章从红曲霉发酵液中分离得到一种被称为monacolink的物质,该物质即为洛伐他汀。由于其疗效显著、毒副作用小、耐受性好而日渐受到重视,是目前在保健食品中唯一应用的他汀类物质。洛伐他汀有两种结构式:酸式和内酯式。酸式洛伐他汀的副作用小,可由内酯式洛伐他汀经过碱催化反应得到。国内外目前对于洛伐他汀的检测方法主要有紫外分光光度法、薄层色谱法和高效液相色谱法。紫外分光光度法和薄层色谱法虽然也能检测出洛伐他汀的含量,但由于样品中其他物质的干扰性太强,会对检验结果产生一定的影响。一般而言,液相色谱法如果方法得当,可以精确的测定其有效含量,但色谱柱、流动相、流速的不同,液相分离的效果亦不同。针对当前文献中所述的洛伐他汀液相检测方法对酸式洛伐他汀无保留,或者难以对酸式洛伐他汀和内酯式洛伐他汀有效分离与检测的情况,本发明提供了一种有效准确测定的方法。
技术实现要素:
本发明的目的在于针对现有技术不足,提供一种准确测定样品中酸式和内酯式洛伐他汀含量的方法。
为实现上述目的,本发明采用如下技术方案:
一种同时测定酸式和内酯式洛伐他汀含量的方法:配置一定浓度梯度的酸式洛伐他汀和内酯式洛伐他汀标准溶液,用液相色谱绘制浓度-峰面积的标准曲线;将样品溶液过0.22μm滤膜后采用液相色谱法进行测定,并根据标准曲线计算浓度。洛伐他汀总量=酸式洛伐他汀含量+内酯式洛伐他汀含量。
(1)酸式洛伐他汀标准溶液的配置方法为:准确称取0.0070g洛伐他汀标品,并将其溶于含有0.8gnaoh的100ml70%(v/v)乙醇中,超声30min,即得浓度为70μg/ml的储备液。分别取0ml、0.2ml、0.4ml、0.6ml、0.8ml、1ml上述储备液加70%(v/v)乙醇溶液定容于1ml,配置成一定浓度梯度的酸式洛伐他汀标准溶液,过0.22μm滤膜后备用。
(2)内酯式洛伐他汀标准溶液的配置方法为:准确称取0.0065g洛伐他汀标品,并将其溶于100ml70%(v/v)乙醇中,即得浓度为65μg/ml的储备液。此过程既不能超声也不能高温,否则内酯式洛伐他汀会部分转化为酸式洛伐他汀。分别取0ml、0.2ml、0.4ml、0.6ml、0.8ml、1ml上述储备液加70%(v/v)乙醇溶液定容于1ml,配置成一定浓度梯度的内酯式洛伐他汀标准溶液,过0.22μm滤膜后备用。
(3)液相色谱条件为:液相色谱柱lunac18(2)4.6×150mm5μ,柱温40℃,紫外237nm,进样量20μl,流动相按体积比为a相(缓冲液):b相(100%乙腈):c相(100%甲醇)=25:5:70,流速1.2ml/min。其中,流动相a(缓冲液)的制备方法为:1ml磷酸、0.2ml三乙胺、0.9g十六烷基三甲基溴化铵,加去离子水定容至1l,过0.45μl膜、超声20min后备用。
本发明的有益效果在于:
本发明所述的方法克服了酸式洛伐他汀无法被色谱柱吸附而被当成“杂质”过早出峰的问题。同时,本发明的方法还能对酸式洛伐他汀和内酯式洛伐他汀很好地分离,并对其准确定量,两峰互不干扰。本发明的方法标曲线性好,两种洛伐他汀的标准曲线r2达0.999以上,对于样品中洛伐他汀的回收率在97%~100%之间,方法准确可行,可信度高。
附图说明
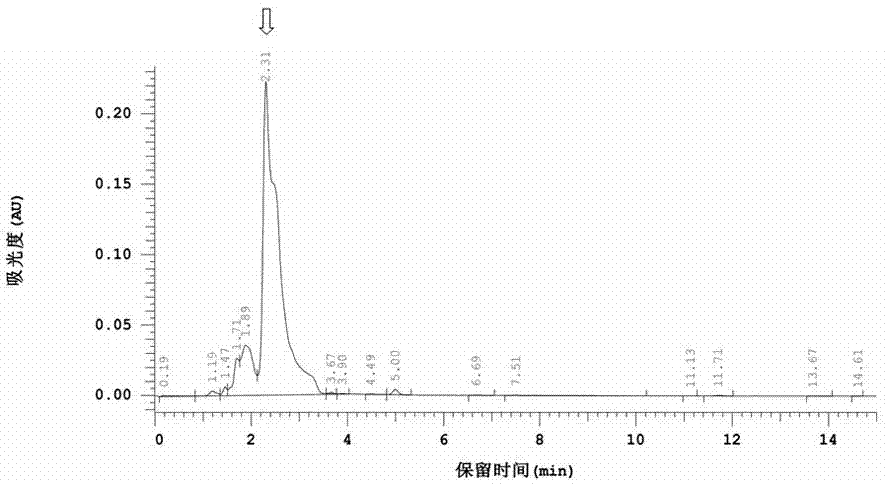
图1为文献中酸式洛伐他汀液相色谱图;
图2为本发明中酸式洛伐他汀液相色谱图;
图3为本发明中内酯式洛伐他汀液相色谱图;
图4为本发明中酸式洛伐他汀的标准曲线;
图5为本发明中内酯式洛伐他汀的标准曲线;
图6为本发明中红曲米乙醇抽提液的液相色谱图。
具体实施方式
以下结合具体实施例对本发明做进一步说明,但本发明不仅仅限于这些实施例。
实施例1
分别采用文献中的方法及本发明的方法对酸式洛伐他汀进行测定。
酸式洛伐他汀溶液的配置:准确称取0.0070g洛伐他汀标品,并将其溶于含有0.8gnaoh的100ml70%(v/v)乙醇中,超声30min,即得浓度为70μg/ml的酸式洛伐他汀溶液。
文献中的方法:液相色谱法:色谱柱:c18150×4.6mm5um,流动相:乙腈:0.1%磷酸=75:25,流速1.0ml/min,进样量20ul,紫外检测器,检测波长237nm。
图1为文献中酸式洛伐他汀液相色谱图,从图中可看出,酸式洛伐他汀的保留时间为2.31min,保留时间过早,色谱柱对样品几乎不保留,因此也会出现目标峰明显与其他杂峰重叠的情况。此方法难以对酸式洛伐他汀进行定性定量测定。
本发明的方法:液相色谱法:色谱柱:c18150×4.6mm5um,紫外237nm,进样量20μl,流动相为a相(缓冲液):b相(100%乙腈):c相(100%甲醇)=25:5:70,流速1.2ml/min。其中,流动相a相的缓冲液为含有1ml磷酸、0.2ml三乙胺、0.9g十六烷基三甲基溴化铵的水溶液。
图2为本发明中酸式洛伐他汀液相色谱图,从图中可以看出,酸式洛伐他汀的保留时间为9.71min,说明该物质与色谱柱之间有吸附、分离的过程。
使用本发明的方法对内酯式洛伐他汀进行测定。内酯式洛伐他汀的配置方法为:准确称取0.0065g洛伐他汀标品,并将其溶于100ml70%(v/v)乙醇中,即得65μg/ml的内酯式洛伐他汀溶液。
图3为本发明中内酯式洛伐他汀液相色谱图。该样品中残留了少量酸式洛伐他汀,酸式洛伐他汀的保留时间为9.95min,内酯式洛伐他汀的保留时间为11.24min,两者能够明显分离,不互相干扰。
对本发明的方法进行标曲的绘制。分别取0ml、0.2ml、0.4ml、0.6ml、0.8ml、1ml70μg/ml酸式洛伐他汀溶液,加70%(v/v)乙醇溶液定容于1ml,配置成一定浓度梯度的标准溶液,过0.22μm滤膜后测定;分别取0ml、0.2ml、0.4ml、0.6ml、0.8ml、1ml65μg/ml内酯式洛伐他汀溶液,加70%(v/v)乙醇溶液定容于1ml,配置成一定浓度梯度的标准溶液,过0.22μm滤膜后测定。
图4和图5分别为酸式洛伐他汀和内酯式洛伐他汀标准曲线。r2分别为0.9996和0.9998,线性好,能够以此为依据对样品进行外标法定量。
使用本发明的方法对红曲米进行洛伐他汀含量的测定。红曲米的样品预处理:用研钵对红曲米进行研磨,取0.0300g红曲米粉末,加20ml70%(v/v)乙醇60℃提取2h后4500r/min离心10min,取上清液过膜后测定。
图6为本发明中红曲米乙醇抽提液的液相色谱图,酸式洛伐他汀和内酯式洛伐他汀的保留时间分别为9.95min、11.24min,两峰互不重叠、互不干扰,说明本发明的方法能对样品中的酸式洛伐他汀和内酯式洛伐他汀进行分离与测定。
以上述红曲米乙醇提取液为样品,对本发明方法进行回收率的测定,测定结果如下:
表1样品加标回收率检测结果
对样品进行加标后,加标回收率在95%~105%范围内则说明方法可信度高,其他因素的干扰小,该方法下的加标回收率均在97%以上,且接近100%,说明本发明的方法准确可行。
综上,新建立的方法能够对酸式洛伐他汀与内酯式洛伐他汀进行分离,分离效果较好,回收率高,方法可行。
- 还没有人留言评论。精彩留言会获得点赞!