使用高效液相色谱法测定利伐沙班及其有关物质的方法与流程
本发明属于分析化学领域,涉及一种使用高效液相色谱法同时测定利伐沙班及其有关物质的方法及应用。
背景技术:
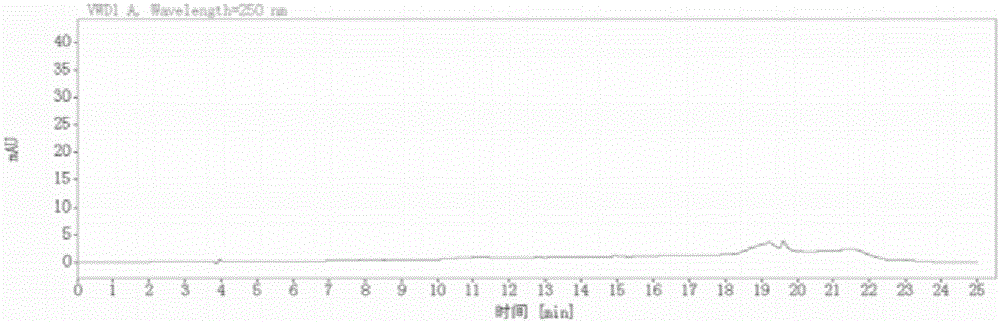
:利伐沙班(rivaroxaban,商品名:xarelto)是由拜耳公司和强生公司联合研制的口服抗凝药物,首次在加拿大上市,是全球第一个口服剂型的直接xa因子抑制剂,至今已在50多个国家陆续上市,市场前景广阔。利伐沙班化学名为:5-氯-氮-[[(5s)-2-氧代-3-[4-(3-氧代吗啉-4-基)苯基]-1,3-噁唑啉-5-基]甲基]噻吩-2-甲酰胺,分子式为c19h18cln3o5s,结构式如下所示:在合成利伐沙班的过程中,一些已知结构的中间体或杂质和未知结构的杂质因去除不完全而残留,会影响利伐沙班的纯度和质量。在药物质量控制中,这些已知结构的中间体或杂质和未知结构的杂质以及利伐沙班的降解产物统称为有关物质,对于这些有关物质,不论在原料药还是其制剂中均需要严格的控制,以保证利伐沙班的产品质量。根据文献报道,目前行业内对于利伐沙班含量及有关物质的检测大都采用国家进口注册标准方法,但是该方法的最大缺点就是无法检测在c18色谱柱上呈现弱保留行为的一些杂质(如本发明化合物列表2中的杂质1、杂质2、杂质3等),并且该方法的空白溶剂干扰主峰和杂质的检测,方法灵敏度低。申请号为“201410746906.8”的发明专利公开了一种测定利伐沙班及其制剂中杂质的方法,所述的流动相a为三氟乙酸与磷酸二氢钠的混合溶液,并明确指出适用ph为4.4-4.6,该发明专利所检测的利伐沙班杂质只包含本发明所述的杂质3、杂质4和杂质7,经试验证明该方法并不能有效分离本发明所述的杂质1、杂质2和杂质3,而杂质1、2、3、4为利伐沙班杂质中最难分离的4个杂质。基于现有技术,对于本发明所述的杂质1、2、3这种电离能力较强不易在反向色谱柱上保留的物质,一般的解决方案是添加离子对试剂以增加其在反向色谱固定相上的保留。专利cn105004802b公布了一种分离测定利伐沙班及其杂质的方法及应用,该方法是在流动相a中加入一定浓度的酸性离子对试剂,来解决在普通c18色谱柱上呈现弱保留行为的一些杂质无法检测这一问题,从而达到分离测定利伐沙班及其杂质的目的。但该方法仍然存在明显缺陷:首先,该方法对流动相ph比较敏感,仅在ph3.7-4.2之间有很好的分离效果(如实施例5中提到的当ph为3.6时,杂质b与g不能有效分离),检测方法耐用性差。并且经实验验证该方法前几个流出的杂质组分峰对称性差,且保留时间重现性不好。其次,该方法仍然不能有效排除空白溶剂对利伐沙班及其有关物质检测的干扰问题,且该专利提到的定量限与检出限实验结果难以重复,实施例中也未给出定量限与检出限的典型图谱;再次,该方法在检测某些杂质(如本发明杂质列表2中的杂质6时),响应值太小,检出限无法达到ich关于杂质的限度检测的一般要求,方法检测灵敏度低。最后,该方法需要在流动相中加入离子对试剂来增强碱性含氮杂质在色谱柱上的保留。而离子对试剂会对色谱柱造成不可逆转的伤害,影响色谱柱固定相上的活性位点,不但影响方法的重现性和耐用性,而且大大地缩短了色谱柱的使用寿命。基于以上原因,现有技术中,对利伐沙班及其有关物质的分离检测,所使用的方法均存在干扰峰影响、耐用性差、重现性差、检测的灵敏度低、并且计算过程中需要手动扣除空白溶剂等问题,对检测结果计算和判定造成很大的影响。当前对步骤简单、灵敏度高、耐用性好、重现性好并且测定结果准确性高的利伐沙班及其有关物质的测定方法仍有迫切需求。技术实现要素:针对现有技术的不足,本发明的目的在于对现有的利伐沙班含量及有关物质检查方法进行改进,并增加了先前技术并未列出的6种杂质的检测,提供一种步骤简单、灵敏度高、耐用性好、重现性好、测定结果准确性高并且分离速度快的利伐沙班及其有关物质的测定方法,对利伐沙班原料药和制剂的质量标准的制定及提高具有重要意义。本发明提供了一种使用高效液相色谱法测定利伐沙班及其有关物质的方法,其中,所述高效液相色谱法包括以下条件:固定相为十八烷基硅烷键合硅胶;流动相a为ph5.0-7.0的磷酸盐缓冲液、枸橼酸盐缓冲液、乙酸盐缓冲液或碳酸盐缓冲液;流动相b为乙腈或甲醇;按下表1进行梯度洗脱;表1梯度洗脱条件时间(分钟)流动相a(体积%)流动相b(体积%)0.00100‐900‐105.00100‐900‐1018.0040‐6060‐4018.10100‐900‐1025.00100‐900‐10优选地,所述流动相a为磷酸二氢钠-磷酸缓冲液;优选地,所述高效液相色谱法的流速为0.2-2.0ml/min;优选地,所述高效液相色谱法采用双波长检测模式,检测波长为220nm,250nm;优选地,所述高效液相色谱法的色谱柱柱温为30-45℃;优选地,对照品溶液与供试品溶液的取样量分别为5-20μl;优选地,所述色谱柱为月旭的lp,其采用特殊的硅烷键合试剂和三位键合工艺,使键合相和硅胶基质的结合度大大高于普通的反向色谱固定相,同时色谱柱端基不封尾,提供了与常规端基封尾的色谱柱不同的色谱选择性。优选地,所述有关物质选自利伐沙班生产中所产生的中间体、杂质或代谢产物;优选地,所述有关物质包括杂质1-杂质8,结构如下表2所示:表2杂质1-杂质8的结构优选地,所述高效液相色谱法包括以下步骤:1)制备利伐沙班及其有关物质的对照品溶液:2)制备供试品溶液;3)高效液相色谱检测;其中,所述高效液相色谱检测的条件如下:色谱柱为月旭lp-c18,规格为250mm*4.6mm,5μm;流动相a为ph为5.0-7.0的磷酸二氢钠-磷酸缓冲液,流动相b为乙腈;对照品溶液与供试品溶液的取样量分别为5-20μl;流速为0.2-2.0ml/min;双波长检测模式,检测波长为220nm,250nm;色谱柱柱温为30-45℃;按表1进行梯度洗脱。其中,梯度洗脱时,可根据实际情况选择流动相b的起始比例在0-10%;所述十八烷基硅烷键合硅胶色谱柱采用特殊的硅烷键合试剂和三位键合工艺,使键合相和硅胶基质的结合度大大高于普通的反向色谱固定相,同时色谱柱端基不封尾,提供了与常规端基封尾的色谱柱不同的色谱选择性。优选地,可根据杂质检测实际情况选择双波长模式同时检测多种微量的利伐沙班杂质。优选地,在对照品溶液与供试品溶液进入高效液相色谱仪的进样器之前,先经过捕集流动相杂质的小柱。优选地,所述步骤1)通过包括以下步骤的方法完成:a.制备流动相a和流动相b;b.制备混合溶剂:将流动相a和流动相b混合,其中流动相a和流动相b的体积比为40-70:60-30,优选地为50:50;c.制备利伐沙班对照品溶液:精密取利伐沙班对照品,置于量瓶中,加入步骤b制备的混合溶剂溶解并定容至刻度,摇匀,作为利伐沙班的对照品溶液;d.制备有关物质对照品溶液:分别精密称取各种有关物质,置于量瓶中,加入步骤b制备的混合溶剂溶解并定容至刻度,作为对照品贮备溶液;然后分别取对照品贮备溶液,置于同一100ml量瓶中,加入混合溶剂溶解并定容至刻度,作为有关物质对照品溶液。优选地,所述步骤2)包括以下步骤:精密称取利伐沙班供试品,置于量瓶中,加入步骤1)操作b制备的混合溶剂溶解并定容至刻度,作为供试品溶液。本发明公开了一种测定利伐沙班及其有关物质的高效液相色谱分析方法,该方法优于其他用于测定利伐沙班及其有关物质的分析方法。本方法选用了一类经过特殊处理的c18色谱柱,结合流动相ph的调整,避免了离子对试剂的使用,使即使电离能力较强、在反向色谱柱上保留很短或根本不保留的杂质也能够很好的分离检测;同时增加了用于流动相杂质捕集的小柱,该小柱安装在进样器之前,不影响样品在色谱系统的洗脱和响应,但能够有效排除空白溶剂对利伐沙班含量及有关物质测定的影响,避免了手动扣除空白溶剂的主观因素影响和软件自动扣除空白溶剂时,由于保留时间误差引起的溶剂扣除失败的情况发生。进一步地,本发明通过对进样量、梯度洗脱程序、配样溶剂等因素的考察,提供了一种专属性好、灵敏度高、重现性好、准确性高、后期数据处理简易的能同时测定利伐沙班含量及有关物质分离检测方法。与现有技术相比,本发明具有以下优点:1)本发明采用了一类经过特殊处理的c18色谱柱,该色谱柱以十八烷基硅烷键合超纯全多孔球形硅胶为固定相,采用特殊的硅烷键合试剂和三位键合工艺使键合相和硅胶基质的结合度大大高于普通的反向色谱固定相,同时色谱柱端基不封尾,避免由于封尾的小基团的水解导致色谱柱性能的不稳定,并提供了与常规端基封尾的色谱柱不同的色谱选择性。2)本发明所使用的检测方法,各杂质组分峰对称性和保留时间重现性更好,并且流动相ph在5.0-7.0之间,利伐沙班及其各杂质间均可达到基线分离,该检测方法耐用性好、测定结果准确度高;3)本发明避免了离子对试剂的使用,使该方法较现有技术有更好的色谱耐用性和重现性,并能避免因离子对试剂的使用而使色谱柱寿命缩短;4)本发明提出了采用双波长检测方法同时测定响应因子差异较大的杂质,以保证检测灵敏度和准确性;通过一次进样即可同时得到全部含量和有关物质分析结果。附图说明以下,结合附图来详细说明本发明的实施方案,其中:图1为使用本发明的方法测定空白对照的hplc图谱;图2为使用本发明的方法测定利伐沙班及杂质1-8的混合样品溶液的hplc图谱图3为现有技术的方法测定利伐沙班及其杂质的空白对照hplc图谱;图4为现有技术的方法测定利伐沙班及其杂质的样品溶液hplc图谱;图5为使用本发明的方法测定利伐沙班粗品的hplc图谱图6为本发明的方法测定利伐沙班片的hplc图谱。具体实施方式以下将参照附图,对本发明的优选实施例进行详细描述。优选实施例中未注明具体条件的试验方法,通常按照常规条件,所举实施例是为了更好地对本发明的内容进行说明,但并不意味着本发明的内容仅限于所举的实施例。熟悉本领域的技术人员根据上述
发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。以下实施例中,采用的仪器及色谱条件如下:高效液相色谱仪:安捷伦1260型号色谱柱:月旭lp-c18(250mm*4.6mm,5μm)杂质捕集小柱:岛津ghosttrapds检测波长:220nm,250nm;流速:1.0ml/min实施例1使用本发明的方法测定利伐沙班含量及其有关物质1.1仪器与条件:安捷伦1260型号的液相色谱系统,色谱柱:月旭lp-c18(250mm*4.6mm,5μm),检测波长为220nm,250nm,色谱柱柱温为35℃,流速1.0ml/min;所述捕集小柱为岛津的ghosttrapds。1.2试验步骤1)流动相a的配制:取1.42g磷酸二氢钠,溶于1000ml水中,用磷酸调节ph至6.0,作为流动相a;流动相b为乙腈;2)制备利伐沙班及其有关物质的对照品溶液:①制备混合溶剂:使用流动相a和流动相b制备混合溶剂,其中流动相a和流动相b的体积比为50:50;②制备利伐沙班对照品溶液:取利伐沙班对照品25mg,精密称定,置于50ml量瓶中,加入混合溶剂溶解并定容至刻度,摇匀,作为利伐沙班的对照品溶液;③有关物质对照品贮备溶液:分别取杂质1、杂质2、杂质3、杂质4、杂质5、杂质6、杂质7、杂质8各25mg,精密称定,置于50ml量瓶中,加入混合溶剂溶解并定容至刻度,摇匀,作为对照品贮备溶液;然后分别取对照品贮备溶液0.150ml,置于同一100ml量瓶中,加入混合溶剂溶解并定容至刻度,摇匀,作为有关物质对照品溶液;3)制备供试品溶液取利伐沙班供试品25mg(供试品由本单位自制,批号161114),精密称定,置于50ml量瓶中,加入步骤2)③中的杂质贮备溶液各0.5ml,用步骤2)①制备的混合溶剂溶解并定容至刻度,摇匀,作为供试品溶液;4)取步骤2)和3)的样品5μl,先经过捕集流动相杂质的小柱,然后进入液相色谱仪;其中,按表3所示进行梯度洗脱:表3梯度洗脱条件时间(分钟)流动相a(%)流动相b(%)0.009555.0095518.00495118.1095525.009555)取流动相a流动相b组成的混合溶剂(50:50,v/v)作为空白对照,另取步骤3)所述供试品溶液,按照以上色谱条件进样,记录色谱图,测定结果见表4,液相色谱图见附图1、附图2。表4供试品溶液检测结果实验结果表明,空白溶剂不干扰供试品的检测;各杂质峰的理论塔板数均大于10000;各色谱峰之间分离度均大于1.5,由此可见,本发明的方法柱效高,分离效果好,空白无干扰。实施例2现有技术检测利伐沙班及其杂质的对比实施例采用专利cn105004802b实施例1所述的色谱柱和检测方法检测利伐沙班的含量及上述的8种有关物质;1)流动相a的配制:称取戊烷磺酸钠2.0g、移取磷酸2.0ml于1000ml容量瓶中,加水溶解并稀释至刻度,用1mol/l的氢氧化钠溶液调节ph至4.0;流动相b为乙腈;2)取利伐沙班25mg,加入本发明实施例1中制备的有关物质对照品贮备溶液适量,用30%乙腈水溶液溶解样品得样品溶液;3)取空白对照和步骤2)的样品溶液各10μl注入岛津lc-20at色谱仪,色谱柱型号为purospherstarrp-18,设置于流动相流速为1.0ml/min,检测波长为250nm,色谱柱柱箱温度为30℃;记录色谱图,样品溶液测定结果见表5,空白对照和样品溶液色谱图见附图3、4;表5样品溶液检测结果实验结果表明,前4个流出的组分在色谱柱上的保留时间非常短,且峰型差、柱效低,方法重现性不好。空白溶剂在9-20min处有干扰峰存在。可以减少干扰。而本发明使用的检测方法大大延长了前4个组分在色谱柱上的保留,不但具有柱效高、峰型好、专属性和重现性好等优势,更重要的是同时避免了离子对试剂的使用,使该方法具有更好的色谱适用性,而且能避免离子对试剂对色谱柱产生的不可逆的损害。实施例3不同流动相ph值对分析检测的影响参照实施例1中方法用磷酸二氢钠溶于水中,分别用磷酸调节ph至6.0、5.5、6.5,作为流动相a;其他条件与实施例1中相同,取实施例1中所述样品溶液注入液相色谱系统进行分析,记录色谱图,检测结果见表6。表6不同流动相ph实施例对比实验结果表明,流动相a的ph分别为6.0、5.5、6.5时,均能很好地分离利伐沙班及其8种有关物质,并且各杂质及利伐沙班的含量结果不受ph变化影响,证明本发明方法有更好的方法耐用性。实施例4不同进样量对分析检测的影响分别取5μl、10μl、20μl样品,按照实施例1的方法将样品注入液相色谱系统进行分析,记录色谱图,检测结果见表7。表7不同进样量实施例对比实验结果表明,当进样量为5-20μl时,前4个流出的组分杂质1、2、3、4的保留时间和分离度不受溶剂效应影响。因此,若要实现同时测定利伐沙班含量及其有关物质检测,在保证含量测定的准确性和精密度同时又要保证有关物质测定时各杂质峰有较好的峰型和分离度,应选择5μl-20μl进样体积。实施例5对利伐沙班及其有关物质的检出限与定量限研究定量限溶液:取实施例1中的有关物质对照品贮备溶液适量,加流动相a和流动相b组成的混合溶剂(50:50,v/v)逐级稀释,得到定量限溶液,检测结果如表8所示。表8定量限检测结果检出限溶液:精密量取定量限溶液,稀释后得检出限溶液,检测结果如表9所示。表9检出限检测结果从上表实验数据可知,本发明的色谱条件下,利伐沙班及其有关物质的检出限与定量限均符合要求。实施例6使用本发明的方法测定利伐沙班含量及其有关物质1.1仪器与条件:岛津lc-20a的液相色谱系统,色谱柱:月旭lp-c18(250mm*4.6mm,5μm),检测波长为250nm,色谱柱柱温为45℃,流速1.3ml/min;1.2试验步骤1)流动相a的配制:取2.1g枸橼酸,溶于1000ml水中,用氢氧化钠调节ph至7.0,作为流动相a;流动相b为乙腈;2)制备利伐沙班及其有关物质的对照品溶液:①制备混合溶剂:使用流动相a和流动相b制备混合溶剂,其中流动相a和流动相b的体积比为40:70;②制备利伐沙班对照品溶液:取利伐沙班对照品25mg,精密称定,置于50ml量瓶中,加入混合溶剂溶解并定容至刻度,摇匀,作为利伐沙班的对照品溶液;3)制备样品溶液取利伐沙班粗品25mg(由本单位自制,批号160822粗品),精密称定,置于50ml量瓶中,加入步骤2)①制备的混合溶剂溶解并定容至刻度,摇匀,作为样品溶液;4)取步骤2)和3)的样品20μl,进入液相色谱仪;其中,按表10所示进行梯度洗脱:表10梯度洗脱条件时间(分钟)流动相a(%)流动相b(%)0.0090105.00901018.00604018.10901025.009010表11样品溶液检测结果实验结果液相色谱图见附图5,该结果表明,在范围内对各参数进行调整后,各杂质峰的理论塔板数和各色谱峰之间分离度均符合要求,可以达到基线分离,由此可见,本发明的方法柱效高,分离效果好,空白无干扰。实施例7利伐沙班片的检测实施例取利伐沙班片5片(10mg/片),置于100ml量瓶中,加流动相a流动相b组成的混合溶剂(50:50,v/v)溶解并定容至刻度,摇匀,过滤,作为样品溶液。取样品溶液,按照实施例1所述方法进样,记录色谱图,液相色谱图见附图6。试验结果表明,色谱图中无干扰峰存在,本发明的方法适用于利伐沙班片剂的质量检测。最后说明,以上实施例仅用于说明本发明的技术方案特点,在本领域的技术人员基于本方案的技术特点进行的修改或等同替换,不脱离本方案的宗旨和范围,均应涵盖在本发明权利要求书当中。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1