氨苄西林和/或氨苄西林钠中特戊酸的检测方法与流程
本发明涉及氨苄西林和/或氨苄西林钠中杂质检测领域,特别是涉及氨苄西林和/或氨苄西林钠中特戊酸的检测方法。
背景技术:
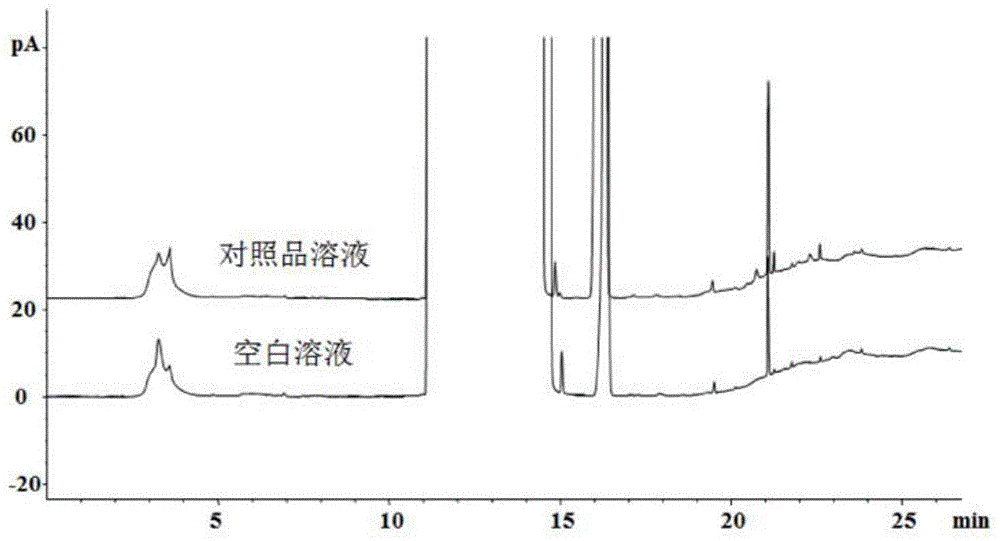
:氨苄西林钠属于青霉素一类的抗生素类药,可以用于肌内注射或者静脉注射。主要用以治疗敏感细菌所致的上、下呼吸道感染、胃肠道感染、尿路感染、皮肤、软组织感染、脑膜炎、败血症、心内膜炎等。氨苄西林作为氨苄西林钠重要的起始物料,根据氨苄西林的生产工艺可知,氨苄西林中可能会有少量的特戊酸残留。为保证氨苄西林钠的安全性和有效性,需对其起始物料氨苄西林中残留的特戊酸进行检测。氨苄西林中残留的特戊酸的测定方法未见国内外文献报道。技术实现要素:本发明提供的氨苄西林和/或氨苄西林钠中杂质的检测方法,采用气相色谱法对待检样品进行检测,根据色谱结果定性或定量,色谱条件包括:色谱柱:以硝基对苯二甲酸改性的聚乙二醇为固定液的毛细管色谱柱或与其相似或同等极性的色谱柱;检测器:fid检测器;柱温升温程序为:初始温度60℃,以20℃/min的速率升至180℃保持5min,再以20℃/min的速率升至200℃,保持3min。所述杂质为合成等工艺过程中引入的杂质,包括反应物、中间体、副产物、试剂、溶剂、催化剂等。进一步地,所述杂质为特戊酸。进一步地,所述色谱柱规格为30m×0.320mm,1.00μm。在本发明的具体实施方式中使用的色谱柱商品名为agilentdb-ffap,但是,只要满足上述描述的色谱柱均可运用于本发明的检测方法,不限于上述商品。进一步地,柱流量为1.0ml/min。进一步地,进样口温度为200℃,进样量为1μl。进一步地,进样方式采取直接进样,进样模式采用分流模式,分流比为1∶1。进一步地,检测器温度为250℃,h2流速为40ml/min,空气流速为400ml/min。特戊酸属于挥发性的高沸点(166℃)有机弱酸,若采用顶空进样,由于顶空中的不锈钢管路较长,对特戊酸中的活性官能团羧基(cooh-)有较强的吸附,致使其检出的灵敏度及低,因此采用直接进样的方式进行特戊酸的检测。当被检测的样品基质为高沸点固体化合物且呈现酸/碱性,同时溶解性较差时,若采用直接进样(仅在dmso中溶解),样品基质由于在现有的色谱柱耐用温度范围内无法气化或气化不完全而残留在色谱仪和柱子内,将对气相色谱仪和色谱柱带来污染及不可逆的损伤,同时无法找到特戊酸的目标峰。此时,样品的前处理(预处理)就变得非常重要。进一步地,所述检测方法包括以下内容:(1)采用液液萃取法对供试品进行前处理;;(2)制备对照品溶液;(3)将对照溶液进样检测;(4)将供试品溶液进样检测。所述检测方法可使用外标法等方法分析、计算检测结果。进一步地,制备供试品溶液或/和对照溶液的步骤为:将样品溶解,用稀释剂稀释到指定浓度后,用低沸点的有机溶剂萃取,取有机相即得。进一步地,所述溶解样品的溶剂和稀释剂为0.2~0.4%(v/v)磷酸水溶液,所述低沸点的有机溶剂选自环己烷、乙醚、异丙醚中的一种或几种。本发明的有益效果是:(1)本发明检测方法能够有效检测氨苄西林和/或氨苄西林钠中特戊酸,为氨苄西林和/或氨苄西林钠中特戊酸的检测提供途径,能够满足工艺过程中的杂质监控及成品中的杂质控制要求。(2)本发明检测方法采用液液萃取法对样品进行前处理,避免了样品基质进入仪器和色谱柱带来的污染,且该方法的操作简单,萃取回收率高(在84.0%~100.0%之间)。(3)本发明检测方法检测特戊酸专属性强、峰形良好(拖尾因子1.0~1.1)、理论塔板数高(大于300000),且方法运行时间较短(一针的运行时间仅为15min),可实现特戊酸的快速分析。(4)本发明检测方法灵敏度高(检测限可达0.0003mg/ml(s/n约6.9),定量限为0.0013mg/ml(s/n约22.7)),准确度(加标回收率在94.05~112.0%之间)和重现性好,结果可靠。附图说明图1是本发明apc-sm1中残留溶剂特戊酸分析方法1叠图;图2是本发明apc-sm1中残留溶剂特戊酸分析方法2叠图;图3是本发明apc-sm1中残留溶剂特戊酸分析方法3叠图;图4是本发明apc-sm1中残留溶剂特戊酸分析方法4叠图;图5是本发明apc-sm1中残留溶剂特戊酸分析方法5叠图;图6是本发明apc-sm1中残留溶剂特戊酸分析方法6叠图。具体实施方式一、仪器设备仪器名称品牌及型号气相色谱仪agilent7890b电子天平xs205du色谱柱agilentdb-ffap(30m*0.320mm,1.00μm)二、试剂和标准品、对照品三、色谱参数条件筛选1.进样方式和色谱柱的选择待测溶剂特戊酸沸点为166℃,选择直接进样方式进行分析;因特戊酸极性较大,选择使用于酸类分析的强极性db-ffap(30m*0.320mm*1.00μm)色谱柱。2.样品前处理方式的筛选2.1直接溶解进样分析根据apc-sm1的溶解性,常规有机溶剂中仅dmso可使供试品溶解,故选dmso为稀释剂;考虑到待测溶剂特戊酸限度较高(0.1%)并结合供试品的溶解性,供试品浓度初步设定为10mg/ml。具体色谱条件如表1:表1apc-sm1中残留特戊酸分析方法色谱条件1结论:如图l所示,限度浓度对照品溶液中特戊酸色谱峰未找到。调整升温程序,其他条件不变,考察方法的专属性,具体条件如表2:表2apc-sm1中残留溶剂特戊酸分析方法色谱条件2结论:如图2所示,调整升温程序后,限度浓度对照品溶液中特戊酸色谱峰仍未找到。2.2萃取后取有机层进样分析发明人通过实验发现,供试品在稀酸或稀碱中溶解性较好而在低沸点的有机溶剂(环己烷、乙醚和异丙醚)中几乎不溶,而特戊酸在低沸点有机溶剂中的溶解性较好。预采用萃取的方式对样品进行前处理,使待测的特戊酸溶于低沸点的有机层,而样品溶于酸水层,再吸取有机层进样分析,该方法既可保证方法的专属性、避免样品进入色谱柱和仪器而带来的污染,又能避免酸水溶液对色谱柱和仪器的腐蚀。3.萃取剂、稀释剂及柱温程序的筛选3.1萃取剂:环己烷;稀释剂:3.34%(v/v)磷酸水溶液;调整升温程序初选3.34%(v/v)磷酸水溶液(1.0ml)溶解供试品(浓度为10mg/ml),用低沸点的环己烷(2.0ml)萃取其中特戊酸后取有机层进行分析,调整升温程序,其他条件不变,考察方法专属性、灵敏度和加标回收率,具体条件如表3:表3apc-sm1中残留溶剂特戊酸分析方法3色谱条件表4apc-sm1中残留溶剂特戊酸分析方法3数据溶剂参考保留时间min拖尾因子理论塔板数回收率%信噪比s/n特戊酸10.0091.233015972.2219.5结论:如图3所示,用环己烷作萃取剂,空白溶液无干扰;如表4所示,采用db-ffap色谱柱,限度浓度对照品溶液中特戊酸峰形良好(拖尾因子为1.2),灵敏度较高(s/n=219.5);但加标供试品溶液中特戊酸回收率为72.2%,且方法重现性较差。预更换萃取剂,并降低磷酸水溶液的浓度,考察特戊酸的萃取回收率和加标回收率。3.2萃取剂:乙醚;稀释剂:0.4%(v/v)磷酸水溶液;调整升温程序将萃取剂更换为乙醚,磷酸水溶液的浓度降为0.4%,考察方法专属性及特戊酸的萃取回收率和加标回收率;特戊酸出峰较早(约10min),可调整升温程序,缩短分析时间,具体色谱条件如表5:表5apc-sm1中残留溶剂特戊酸分析方法色谱条件4表6apc-sm1中残留溶剂特戊酸分析方法4数据溶剂保留时间min拖尾因子理论塔板数分离度萃取回收率%信噪比s/n特戊酸8.2291.32465864.4116.9287.8结论:如图4所示,用0.4%磷酸水溶液(2.0ml)溶解样品,乙醚(2.0ml)作萃取剂,调整升温程序后,总运行时间为15min,较之前缩短了3min;空白溶液无干扰;如表6所示,专属性溶液中其他溶剂均不干扰特戊酸的检测,特戊酸与其前后相邻峰的分离度为4.4;限度浓度对照品溶液中特戊酸的信噪比s/n为187.8,灵敏度良好;但特戊酸的萃取回收率为116.9%,故需继续优化萃取剂,考察萃取回收率。3.3萃取剂:异丙醚;稀释剂:0.4%(v/v)磷酸水溶液更换异丙醚作萃取剂,色谱条件不变(同表5),考察特戊酸的萃取率及加标回收率。表7apc-sm1中残留溶剂(特戊酸)分析方法5数据结论:如图5所示,用异丙醚(2.0ml)作萃取剂,空白溶液无干扰;专属性溶液中特戊酸与其前后相邻色谱峰的分离度为4.3,其他溶剂均不干扰特戊酸的检测;如表7所示,100%限度浓度对照品溶液中特戊酸萃取回收率为91.2%,拖尾因子在1.4~1.5之间,理论塔板数大于5000,信噪比s/n为184.2,灵敏度和峰形良好;供试品溶液中无杂质峰干扰特戊酸的检测;100%和150%的加标供试品溶液中特戊酸的回收率良好,但50%加标供试品溶液中特戊酸回收率偏低(89.5%),预再次降低磷酸水溶液的浓度,考察特戊酸的加标回收率。3.4萃取剂:异丙醚;稀释剂:0.2%(v/v)磷酸水溶液将磷酸水溶液的浓度降为0.2%,用5.0ml0.2%磷酸水溶液使10mg供试品完全溶解,再加2.0ml异丙醚萃取其中的特戊酸,其余色谱条件同表5,考察特戊酸的加标回收率。表8apc-sm1中残留溶剂特戊酸分析方法6数据结论:如表8所示,将磷酸水溶液的浓度降为0.2%后,三个浓度水平(50%、100%、150%)的限度浓度对照品溶液中特戊酸的萃取回收率(均在94.2%~94.5%之间)及加标供试品溶液中特戊酸的回收率(均在98.0%~105.0%之间)均良好;限度浓度对照品溶液中特戊酸的峰形好(拖尾因子为1.5)、灵敏度高(s/n=252.2)。如图6所示,apc-sm1中可能残留的其他溶剂均不干扰特戊酸的检测。综上,该方法适用于apc-sm1中特戊酸的检测。表9apc-sm1中残留溶剂特戊酸分析方法筛选小结4.最佳色谱条件对照品溶液:取特戊酸适量,用0.2%(v/v)磷酸水溶液溶解并定量稀释成每1ml中约含有特戊酸0.004mg的溶液,作为对照品溶液①,精密量取对照品溶液①5.0ml置具塞试管中,加入2.0ml异丙醚,剧烈振摇,静置分层,取上层液,作为对照品溶液。供试品溶液:取供试品约20mg,精密称定,置具塞试管中,加入5.0ml0.2%(v/v)磷酸水溶液,振摇使溶解,再加入2.0ml异丙醚,剧烈振摇,静置分层,取上层液,作为供试品溶液。表10最佳色谱参数和计算方式四、方法验证程序及验证结果表11apc-sm1残留溶剂特戊酸分析方法验证结果小结以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的
技术领域:
,均同理包括在本发明的专利保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1