一种中心切割二维色谱质谱联用分析烟草中农残的方法与流程
本发明属于烟草中农药残留量分析的技术领域,涉及一种中心切割二维色谱质谱联用分析烟草中农残的方法,具体涉及一种运用中心切割第一维体积排阻液相色谱与第二维反相液相色谱的多维色谱/串联质谱联用分析烟草中多种农药残留量的方法。
背景技术:
烟草作为世界上种植最广泛的非粮食作物,在全世界129个国家和地区种植。因此,农药残留作为烟草质量安全问题的一个重要组成部分,农药残留的测定一直是研究热点。烟草农残常用的方法主要有gc-ms/ms和lc-ms/ms方法。而基质的干扰一直是农药残留量准确测定的重要难题,现有技术有运用液液萃取、固相萃取柱(spe)等技术净化样品,运用gc分离后进入质谱测定,从而达到减少基质效应的效果。还有采用低温和分散微萃取技术净化样品,后并进行lc-ms/ms测定。但是烟草农残检测过程中存在一些大分子的蛋白及一些非极性物质子基质难以去除,基质抑制效应较为明显的问题,影响分析的准确性和重复性。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种中心切割二维色谱质谱联用分析烟草中农残的方法,用于同时测定烟草中多种农药残留且减少烟草农残检测中的基质效应问题。
为实现上述目的及其他相关目的,本发明第一方面提供一种中心切割二维色谱质谱联用分析烟草中农残的方法,对烟草进行样品前处理,采用中心切割二维色谱质谱联用系统对前处理所得待测样品进行定性定量分析,所述中心切割二维色谱质谱联用系统包括沿样品进样方向依次连接的第一维色谱、分流器、定量环、第二维色谱、质谱。
较佳地,所述烟草中农残选自啶虫脒、马拉硫磷、阿拉酸式苯-s-甲基、灭蚜磷、涕灭威砜、甲霜灵、嘧菌酯、甲硫威、苯霜灵、灭虫威砜、双苯三唑醇、灭虫威亚砜、除草定、速灭磷、仲丁灵、久效磷、甲萘威、腈菌唑、多菌灵、敌草胺、壳百威、氧乐果、氯虫苯甲酰胺、噁霜灵、毒虫畏、克草敌、毒死蜱、戊菌唑、甲基内吸磷、二甲戊灵、二嗪磷、磷胺、乙霉威、抗蚜威、甲氟磷、丙溴磷、乐果、霜霉威、烯酰吗啉、残杀威、双苯酰草胺、吡蚜酮、乙拌磷亚砜、吡菌磷、乙硫磷、喹硫磷、灭线磷、精喹禾灵、咪唑菌酮、特丁硫磷、苯线磷、特丁硫磷砜、苯线磷砜、特丁硫磷亚砜、苯线磷亚砜、杀虫畏、丰索磷、噻虫嗪、倍硫磷亚砜、硫双威、氟吗啉、甲基硫菌灵、庚烯磷、三唑酮、吡虫啉、三唑醇、茚虫威、三唑磷、异稻瘟净、杀铃脲、氯唑磷、烯效唑、稻瘟灵、蚜灭磷中的任意一种或多种组合。所述烟草中农残共74种。
较佳地,所述样品前处理包括以下步骤:
1)将烟草加水振荡浸润后静置,再加入内标溶液进行第一次涡旋振荡;
2)再加入试剂盒粉包进行第二次涡旋振荡后第一次离心,取第一上清液;
3)将第一上清液放入试剂盒含吸附剂小管中,进行第三次涡旋振荡后第二次离心,取第二上清液过滤后,即得待测样品。
优选地,步骤1)中,所述烟草为烟草粉末。
优选地,步骤1)中,所述水为纯水。
优选地,步骤1)中,所述烟草加入的质量g与水加入的体积ml之比为2:5-15。更优选地,所述烟草加入的质量g与水加入的体积ml之比为2:10。
优选地,步骤1)中,所述静置时间为8-12min。更优选地,所述静置时间为10min。
优选地,步骤1)中,所述烟草加入的质量g与内标溶液加入的体积ml之比为2:5-15。更优选地,所述烟草加入的质量g与内标溶液加入的体积ml之比为2:10。
优选地,步骤1)中,所述内标溶液为含内标样品的乙腈溶液。所述内标溶液即分别称取内标样品加入乙腈定容后获得。
更优选地,所述内标样品为三磷酸苯酯(tpp)。
进一步优选地,所述内标样品的浓度为100ng/ml。
优选地,步骤1)中,所述第一次涡旋振荡的条件为:转速为1500-2500r/min;时间为0.8-1.2min。更优选地,所述第一次涡旋振荡的条件为:转速为2000r/min;时间为1.0min。
优选地,步骤2)或3)中,所述试剂盒为quechers(quick、easy、cheap、effective、rugged、safe)试剂盒。
优选地,步骤2)中,所述试剂盒粉包内包括有无水硫酸镁、氯化钠、柠檬酸氢钠。
更优选地,所述无水硫酸镁、氯化钠、柠檬酸氢钠与烟草加入的质量之比为4:1:0.5:1.5-2.5。进一步优选地,所述无水硫酸镁、氯化钠、柠檬酸氢钠与烟草加入的质量之比为4:1:0.5:2。
优选地,步骤2)中,所述第二次涡旋振荡的条件为:转速为1500-2500r/min;时间为1.8-2.2min。更优选地,所述第二次涡旋振荡的条件为:转速为2000r/min;时间为2.0min。
优选地,步骤2)中,所述第一次离心的条件为:转速为4500-5500r/min;时间为9-11min。更优选地,所述第一次离心的条件为:转速为5000r/min;时间为10min。
优选地,步骤3)中,所述吸附剂小管内包括有无水硫酸镁和n-丙基乙二胺。所述n-丙基乙二胺为键合固定相吸附剂。
更优选地,所述无水硫酸镁、n-丙基乙二胺加入的质量mg与第一上清液加入体积ml之比为150:25:0.5-1.5。
进一步优选地,所述无水硫酸镁、n-丙基乙二胺加入的质量mg与第一上清液加入体积ml之比为150:25:1.0。
优选地,步骤3)中,所述第三次涡旋振荡的条件为:转速为1500-2500r/min;时间为1.8-2.2min。更优选地,所述第二次涡旋振荡的条件为:转速为2000r/min;时间为2.0min。
优选地,步骤1)、2)、3)中,所述涡旋振荡采用涡旋振荡仪进行。
优选地,步骤3)中,所述第二次离心的条件为:转速为4500-5500r/min;时间为4-6min。更优选地,所述第二次离心的条件为:转速为5000r/min;时间为5min。
优选地,步骤3)中,所述过滤为有机相滤膜过滤的方式。更优选地,所述有机相滤膜的孔径为0.22μm。
较佳地,所述采用中心切割二维色谱质谱联用系统对前处理所得待测样品进行定性定量分析,包括以下步骤:
a)配制标准样品:分别取烟草中农残成分的标样,加入内标样品,再加入乙腈定容,配成混合标准溶液;
b)样品定性检测:分别将步骤a)配制的标准样品和经样品前处理后待测样品进行中心切割二维色谱质谱联用系统检测,通过第一维色谱进行馏分后,馏分目标采用分流器分流、定量环收集,再由第二维色谱进行分离,通过质谱进行测定,比较保留时间、质谱扫描进行定性,确定待测样品中农残成分;
c)样品定量检测:分别将步骤a)配制的标准样品和经样品前处理后待测样品进行中心切割二维色谱质谱联用系统检测,通过第一维色谱进行馏分后,馏分目标采用分流器分流、定量环收集,再由第二维色谱进行分离,通过质谱进行测定,采用内标标准曲线法进行mrm定量,获得待测样品中农残成分的含量。
优选地,步骤a)中,所述混合标准溶液中农残成分的浓度均为10~500ng/ml。
优选地,步骤a)中,所述内标样品为三磷酸苯酯(tpp)。
更优选地,所述内标样品的浓度为100ng/ml。
优选地,步骤b)或c)中,所述第一维色谱为体积排阻液相色谱(gpc)。即采用液相色谱仪进行体积排阻(也称凝胶过滤)。
优选地,步骤b)或c)中,所述第一维色谱包括有依次连接的第一泵、第一分离柱、紫外检测器。
更优选地,所述第一泵与第一分离柱之间设有进样阀。
进一步优选地,所述进样阀为二位六通阀,所述进样阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第4接口为进样口,所述第3接口为排废口,所述第1接口经管线与第一泵相连通,所述第6接口经管线与第一分离柱相连通。
更优选地,所述第一泵为常规使用的液相色谱仪的专用泵。所述第一泵为四元泵。
更优选地,所述紫外检测器为常规使用的液相色谱仪的紫外检测器。
优选地,步骤b)或c)中,所述第一维色谱的条件为:第一分离柱为supersw型凝胶色谱柱;紫外检测器的波长为220-240nm;柱温:25-35℃;流速:0.3-0.5ml/min;进样量:2-4μl;第一维流动相:30-50%乙腈。
更优选地,所述第一维液相色谱的条件为:第一分离柱为tosohtsk-gelsupersw3000型凝胶色谱柱;紫外检测器的波长为230nm;柱温:30℃;流速:0.4ml/min;进样量:3μl;第一维流动相:40%乙腈。
优选地,步骤b)或c)中,所述分流器的分流比为3-10:1。更优选地,所述分流器的分流比为7:1。所述分流器为常规使用的分流器。
优选地,所述步骤b)或c)中,所述馏分目标在定量环收集后要采用中心切割法,获得农残成分的馏分。
所述中心切割法为多维色谱中常用的分离方法,在本发明的二维色谱中,即将第一维色谱分离出的目标物馏分的色谱峰的切割完整转移至第二维色谱,其它非目标物馏分不进第二维色谱。
更优选地,所述中心切割法的切割时间为8-14min。
优选地,步骤b)或c)中,所述定量环的容积为490-510μl;所述定量环的内径为0.95-1.05mm。更优选地,所述定量环的容积为500μl;所述定量环的内径为1.00mm。所述定量环为常规使用的定量环(loop环)。
优选地,步骤b)或c)中,所述第二维色谱为反相液相色谱(rplc)。即采用液相色谱仪进行反相液相分离。
优选地,步骤b)或c)中,所述第二维色谱包括有依次连接的第二泵、第二分离柱。
更优选地,所述第二泵为常规使用的液相色谱仪的专用泵。所述第二泵为四元泵。
更优选地,所述第二泵与第二分离柱之间设有连通阀。
更优选地,所述连通阀为二位六通阀,所述连通阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第1接口和第4接口分别经管线与定量环的两端相连通,所述第2接口经管线与第二分离柱相连通,所述第3接口经管线与第二泵相连通,所述第5接口为排废口,所述第6接口经管线与分流器相连通。
优选地,步骤b)或c)中,所述第二维色谱的条件为:第二分离柱为c18色谱柱;柱温:25-35℃;流速:0.3-0.5ml/min;进样量:2-4μl;第二维流动相a相:1-3mmol/l甲酸铵、0.05-0.15%甲酸、94-96%h2o+4-6%ch3cn(v:v);第二维流动相b相:1-3mmol/l甲酸铵、0.05-0.15%甲酸、4-6%h2o+94-96%ch3cn(v:v);梯度洗脱。
更优选地,所述第二维液相色谱的条件为:第二分离柱为thermoscientificacclaimpolaradvantageⅱc18(4.6*50mm,3μm)色谱柱;柱温:30℃;流速:0.4ml/min;进样量:3μl;第二维流动相a相:2mmol/l甲酸铵、0.1%甲酸、95%h2o+5%ch3cn(v:v);第二维流动相b相:2mmol/l甲酸铵、0.1%甲酸、5%h2o+95%ch3cn(v:v);梯度洗脱。
上述百分比%为体积百分比。
进一步优选地,所述梯度洗脱的具体程序为:
0-17min,a相:b相体积比为95:5-95:5;
17-20min,a相:b相体积比为95:5-60:40;
20-44min,a相:b相体积比为60:40-20:80;
44-46min,a相:b相体积比为20:80-0:100;
46-56min,a相:b相体积比为0:100-0:100;
56-57min,a相:b相体积比为0:100-95:5;
57-60min,a相:b相体积比为95:5-95:5。
优选地,所述步骤b)或c)中,所述质谱的条件为:
质谱:三重四极杆质谱;电离方式:电喷雾离子源(esi);扫描方式:正、负离子扫描,多反应监测(mrm);喷雾(is)电压为:5490-5510v;雾化气(gas1)压力为:413-415kpa;气帘气(curtaingas)压力为:206.5-206.9kpa;辅助雾化气(gas2)压力为:482.4-482.8kpa;离子源温度(tem)为:495-505℃。
更优选地,所述质谱的条件为:
质谱:agilenttechnologies6460triplequadms三重四极杆质谱;电离方式:电喷雾离子源(esi);扫描方式:正、负离子扫描,多反应监测(mrm);喷雾(is)电压为:5500v;雾化气(gas1)压力为:414kpa;气帘气(curtaingas)压力为:206.7kpa;辅助雾化气(gas2)压力为:482.6kpa;离子源温度(tem)为:500℃。
优选地,所述步骤c)中,所述内标标准曲线法包括以下步骤:
所述内标标准曲线法是指在标准溶液中加入内标物质,进行仪器测定,获得标准工作曲线,进而测定含有内标物质的实际样品浓度的方法。
ⅰ、将步骤a)的一系列不同浓度的混合标准溶液分别进行中心切割二维色谱质谱联用系统检测,分别获得多种农残成分/相应内标物的二级选择离子峰面积比与多种农残成分/相应内标物的质量浓度比的线性关系,绘制相应的标准工作曲线,分别计算得到多种农残成分标准工作曲线的回归方程。
进一步的,所述标准曲线中,以多种农残成分与相应内标物的二级选择离子峰面积比为纵坐标(y轴),其多种农残成分与相应内标物的质量浓度比为横坐标(x轴)。
ⅱ、将经样品前处理后待测样品溶液,进行中心切割二维色谱质谱联用系统检测,将获得的待测液中多种农残成分/相应内标物的二级选择离子峰面积比,代入步骤ⅰ中相应的多种农残成分标准工作曲线的回归方程,并根据加入相应内标物的已知质量浓度,计算得到待测样品溶液中多种农残成分的质量浓度。
本发明第二方面提供一种中心切割二维色谱质谱联用系统,包括有第一维色谱、分流器、定量环、第二维色谱、质谱;所述第一维色谱包括有第一泵、第一分离柱、紫外检测器,所述第一泵与第一分离柱之间设有进样阀;所述第二维色谱包括有第二泵、第二分离柱,所述第二泵与第二分离柱之间设有连通阀;所述进样阀分别经管线与所述第一泵、第一分离柱的进样端相连通,所述第一分离柱的出样端、紫外检测器、分流器沿样品进样方向依次连通,所述连通阀分别经管线与分流器、定量环、第二泵、第二分离柱的进样端相连通,所述第二分离柱的出样端与质谱相连通。
较佳地,所述进样阀为二位六通阀,所述进样阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第4接口为进样口,所述第3接口为排废口,所述第1接口经管线与第一泵相连通,所述第6接口经管线与第一分离柱相连通。
较佳地,所述连通阀为二位六通阀,所述连通阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第1接口和第4接口分别经管线与定量环的两端相连通,所述第2接口经管线与第二分离柱相连通,所述第3接口经管线与第二泵相连通,所述第5接口为排废口,所述第6接口经管线与分流器相连通。
所述中心切割二维色谱质谱系统中的第一维色谱、进样阀、分流器、定量环、连通阀、第二维色谱要相匹配。所述匹配是指上述各部件经连通后作为一个整体使用时,能够有效运行。
本发明第三方面提供一种中心切割二维色谱质谱联用系统的使用方法,包括以下步骤:
a)打开第一维色谱和进样阀,第一维流动相通过第一泵由进样阀的第1接口进入,同时待测样品溶液由进样阀的第4接口进入,经第5接口,与第一维流动相由第6接口经管线进入第一分离柱进行馏分,并经紫外检测器检测;
b)多余的第一维流动相,由进样阀的第1接口切换经第2接口至第3接口经管线排出;
c)将步骤1)中馏分目标物经分流器分流后,打开连通阀,将分流物经管线由连通阀的第6接口进入,流到第1接口,经管线进入定量环收集;
d)切换连通阀,打开第二维色谱,通过第二泵由第3接口经第4接口反冲定量环,使定量环收集的待测物由第1接口经第2接口进入第二分离柱进行分离,再经管线进入质谱进行测定;
e)测定后,切换连通阀,使定量环内的多余待测物由第4接口至第5接口经管线排出。
较佳的,步骤a)中,所述紫外检测器用于防止待测成分损失。
如上所述,本发明提供的一种中心切割二维色谱质谱联用分析烟草中农残的方法,运用中心切割第一维体积排阻液相色谱与第二维反相液相色谱的二维色谱/串联质谱联用法测定烟草中74种农残成分,即首先通过一维的体积排阻液相色谱中填料孔径,对烟草萃取液中大分子物质进行分离去除;为解决gpc流动相对第二维反相lc分离的影响,避免中心切割过程中gpc柱压力过高,目标馏分经分流后切割至定量环收集,目标物收集完成时将仅允许少部分目标物通过反冲定量环带入第二维反相液相色谱进一步分离,最后进入质谱mrm定量。具有以下有益效果:
(1)本发明提供的一种中心切割二维色谱质谱联用分析烟草中农残的方法,根据体积排阻液相色谱(gpc)以填料孔径按分子大小分离待测样,能够有效净化去除大分子干扰物质;再根据反相液相色谱(rplc)所具有的高效分离能力,从而降低基质影响,减少离子源污染,保持大批量样品农残特别是大样本量农残测试的重复性、稳定性和准确性。
(2)本发明提供的一种中心切割二维色谱质谱联用分析烟草中农残的方法,能够对74种农残定性定量分析,农药的回收率大多在72~136%之间,定量限在0.022~0.152mg/kg之间。
(3)本发明提供的一种中心切割二维色谱质谱联用分析烟草中农残的方法,其检测限、定量限可以满足日常检测需求,该方法仪器自动运行,可用于烟草农残长时间、大样品量、多组分分析检测。
(4)本发明提供的一种中心切割二维色谱质谱联用分析烟草中农残的方法,不仅可以用于烟草多农残测定中大分子物质的去除,在复杂基质样品中大分子去除小分子目标物分析中亦具有广阔的应用前景。
附图说明
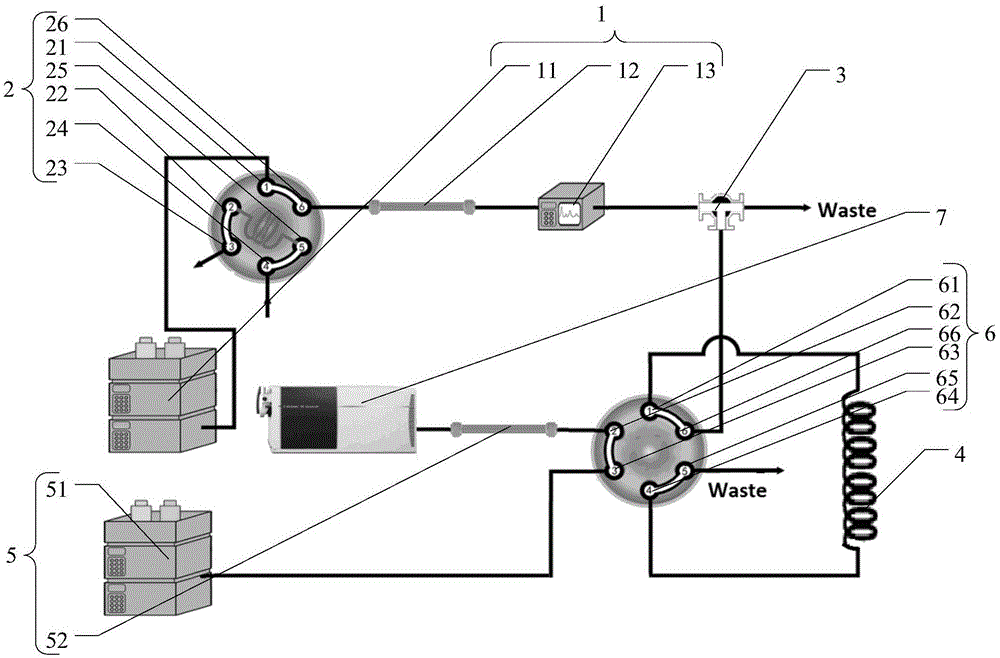
图1显示为本发明中的gpc-rplc串联质谱联用装置的结构示意图。
图中,
1、第一维色谱,
11、第一泵,
12、第一分离柱,
13、紫外检测器,
2、进样阀,
21、第1接口,
22、第2接口,
23、第3接口,
24、第4接口,
25、第5接口,
26、第6接口,
3、分流器,
4、定量环,
5、第二维色谱,
51、第二泵,
52、第二分离柱,
6、连通阀,
61、第1接口,
62、第2接口,
63、第3接口,
64、第4接口,
65、第5接口,
66、第6接口,
7、质谱。
图2显示为本发明中的加标萃取液与纯标样一维分离后质谱总离子流图。
图3显示为lc-ms/ms方法测定烟草农残监控标样的质谱总离子流图。
图4显示为本发明中的gpc-rplc串联质谱法测定烟草农残监控标样的质谱总离子流图。
具体实施方式
下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
以下实施例中使用的试剂及实验用具均为常规使用的试剂及实验用具,均可从市场上购买获得。具体使用试剂和仪器如下:
1、试剂
空白烟叶样品,为0.42mm(40目)烤烟烟末,购自英国食品分析能力评价体系(foodanalysisperformanceassessmentscheme,fapas);农药残留标样纯度大于99.8%、浓度1mg/ml,购自百灵威公司;三磷酸苯酯(tpp)纯度大于99.8%、浓度1mg/ml,购自百灵威公司;quechers试剂盒(粉包含4g无水硫酸镁、1g氯化钠和0.5g柠檬酸氢钠,小管含150mg无水硫酸镁和25mgn-丙基乙二胺键合固定相吸附剂),购自百灵威公司;乙腈、甲酸均为色谱纯,购自德国merk公司;过针孔滤膜为0.22μm的有机相滤膜,购自安谱公司;甲酸铵为分析纯,购自国药集团化学试剂有限公司。
2、仪器
第一维色谱和第二维色谱为agilent1290液相色谱仪,美国agilent公司;六通阀,美国agilent公司;absciex三重四级杆质谱仪api4000,美国absciex公司;电子天平,感量0.0001g,瑞士mettlertoledo公司;sw12h超声仪,瑞士sonoswiss公司;talboysadvancedmulti-tubevortexer涡旋振荡仪,美国henrytroemner.llc公司;eppendorfcentrifuge5810r离心机,德国eppendorfag公司;milli-q纯水仪,美国millpore公司;gpc色谱柱:tosohtsk-gelsupersw3000(4.6*300mm,4μm),安谱公司;反相液相色谱柱:welchultimatexbc18(2.1*200mm,3μm),安谱公司。
如图1所示,本发明中的一种中心切割二维色谱质谱联用系统,包括有第一维色谱、分流器、定量环、第二维色谱、质谱;所述第一维色谱包括有第一泵、第一分离柱、紫外检测器,所述第一泵与第一分离柱之间设有进样阀;所述第二维色谱包括有第二泵、第二分离柱,所述第二泵与第二分离柱之间设有连通阀;所述进样阀分别经管线与所述第一泵、第一分离柱的进样端相连通,所述第一分离柱的出样端、紫外检测器、分流器沿样品进样方向依次连通,所述连通阀分别经管线与分流器、定量环、第二泵、第二分离柱的进样端相连通,所述第二分离柱的出样端与质谱相连通。
实施例1
1、样品前处理
取一定量的烟草粉末样品,加入纯水振荡至样品被水充分浸润后静置8-12min,烟草加入的质量g与水加入的体积ml之比为2:5-15。再加入内标溶液,置于涡旋振荡仪以1500-2500r/min振荡0.8-1.2min。内标溶液为含内标样品的乙腈溶液,内标溶液中内标样品的浓度均为100ng/ml,烟草加入的质量g与内标溶液加入的体积ml之比为2:5-15。
再加入quechers(quick、easy、cheap、effective、rugged、safe)试剂盒粉包,置于涡旋振荡仪以1500-2500r/min振荡1.8-2.2min,再以4500-5500r/min的速度离心9-11min,取第一上清液。试剂盒粉包内包括有无水硫酸镁、氯化钠、柠檬酸氢钠,无水硫酸镁、氯化钠、柠檬酸氢钠与烟草加入的质量之比为4:1:0.5:1.5-2.5。
将第一上清液放入quechers试剂盒含吸附剂小管中,用涡旋振荡仪以1500-2500r/min振荡1.8-2.2min,再以4500-5500r/min的速度离心4-6min,取第二上清液用0.22μm的有机相滤膜过滤后,即得待测样品。吸附剂小管内包括有无水硫酸镁和n-丙基乙二胺。无水硫酸镁、n-丙基乙二胺加入的质量mg与第一上清液加入体积ml之比为150:25:0.5-1.5。
2、标准溶液的配制
分别取烟草中农残成分的标样,加入内标样品,再加入乙腈定容,配成混合标准溶液,混合标准溶液中农残成分的浓度均为10~500ng/ml,内标样品为三磷酸苯酯,内标样品的浓度为100ng/ml。
3、样品含量的测定
分别将2配制的标准样品和经样品前处理后待测样品进行中心切割二维色谱质谱联用系统检测,通过第一维色谱进行馏分后,馏分目标采用分流器分流、定量环收集,再由第二维色谱进行分离,通过质谱进行测定,比较保留时间、质谱扫描进行定性,确定待测样品中农残成分;同时采用内标标准曲线法进行mrm定量,获得待测样品中农残成分的含量。
具体来说,内标标准曲线法是先将上述2中一系列不同浓度的混合标准溶液分别进行中心切割二维色谱质谱联用系统检测,分别获得多种农残成分/相应内标物的二级选择离子峰面积比与多种农残成分/相应内标物的质量浓度比的线性关系,绘制相应的标准工作曲线,以多种农残成分与相应内标物的二级选择离子峰面积比为纵坐标(y轴),其多种农残成分与相应内标物的质量浓度比为横坐标(x轴),分别计算得到多种农残成分标准工作曲线的回归方程。
再将上述1中经样品前处理后待测样品溶液,进行中心切割二维色谱质谱联用系统检测,将获得的待测液中多种农残成分/相应内标物的二级选择离子峰面积比,代入步骤ⅰ中相应的多种农残成分标准工作曲线的回归方程,并根据加入相应内标物的已知质量浓度,计算得到待测样品溶液中多种农残成分的质量浓度。
其中,第一维色谱为体积排阻液相色谱(gpc)。第一维色谱的条件为:第一分离柱为supersw型凝胶色谱柱;紫外检测器的波长为220-240nm;柱温:25-35℃;流速:0.3-0.5ml/min;进样量:2-4μl;第一维流动相:30-50%乙腈。
分流器的分流比为3-10:1。馏分目标在定量环收集后要采用中心切割法,获得农残成分的馏分。即将第一维色谱分离出的目标物馏分的色谱峰的切割完整转移至第二维色谱,其它非目标物馏分不进第二维色谱。如图2所示,通过对第一维色谱的馏分收集并离线分析,发现大分子干扰成分流出主要集中在4-7min。将第一维色谱分离后样品直接引入质谱,进行多反应监测模式扫描,发现农残目标物集中在9-13min,因此中心切割法的切割时间为8-14min。
第二维色谱为反相液相色谱(rplc)。第二维色谱的条件为:第二分离柱为c18色谱柱;柱温:25-35℃;流速:0.3-0.5ml/min;进样量:2-4μl;第二维流动相a相:1-3mmol/l甲酸铵、0.05-0.15%甲酸、94-96%h2o+4-6%ch3cn(v:v);第二维流动相b相:1-3mmol/l甲酸铵、0.05-0.15%甲酸、4-6%h2o+94-96%ch3cn(v:v);梯度洗脱。
梯度洗脱的具体程序为:
0-17min,a相:b相体积比为95:5-95:5;
17-20min,a相:b相体积比为95:5-60:40;
20-44min,a相:b相体积比为60:40-20:80;
44-46min,a相:b相体积比为20:80-0:100;
46-56min,a相:b相体积比为0:100-0:100;
56-57min,a相:b相体积比为0:100-95:5;
57-60min,a相:b相体积比为95:5-95:5。
质谱的条件为:
质谱:三重四极杆质谱;电离方式:电喷雾离子源(esi);扫描方式:正、负离子扫描,多反应监测(mrm);喷雾(is)电压为:5490-5510v;雾化气(gas1)压力为:413-415kpa;气帘气(curtaingas)压力为:206.5-206.9kpa;辅助雾化气(gas2)压力为:482.4-482.8kpa;离子源温度(tem)为:495-505℃。
另外,本发明的中心切割二维色谱质谱联用系统在具体测定时,如图1所示,第一维色谱包括有依次连接的第一泵、第一分离柱、紫外检测器。第一泵与第一分离柱之间设有进样阀。
如图1所示,所述进样阀为二位六通阀,所述进样阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第4接口为进样口,所述第3接口为排废口,所述第1接口经管线与第一泵相连通,所述第6接口经管线与第一分离柱相连通。
如图1所示,所述第一泵为常规使用的液相色谱仪的专用泵。所述第一泵为四元泵。所述紫外检测器为常规使用的液相色谱仪的紫外检测器。
如图1所示,所述分流器为常规使用的分流器。所述定量环为常规使用的定量环(loop环)。所述定量环的容积为490-510μl;所述定量环的内径为0.95-1.05mm。
如图1所示,第二维色谱包括有依次连接的第二泵、第二分离柱。第二泵与第二分离柱之间设有连通阀。
如图1所示,所述连通阀为二位六通阀,所述连通阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第1接口和第4接口分别经管线与定量环的两端相连通,所述第2接口经管线与第二分离柱相连通,所述第3接口经管线与第二泵相连通,所述第5接口为排废口,所述第6接口经管线与分流器相连通。
如图1所示,所述第二泵为常规使用的液相色谱仪的专用泵。所述第二泵为四元泵。
如图1所示,采用中心切割二维色谱质谱联用系统进行测定时,打开第一维色谱和进样阀,第一维流动相通过第一泵由进样阀的第1接口进入,同时待测样品溶液由进样阀的第4接口进入,经第5接口,与第一维流动相由第6接口经管线进入第一分离柱进行馏分,并经紫外检测器检测。多余的第一维流动相,由进样阀的第1接口切换经第2接口至第3接口经管线排出。将馏分目标物经分流器分流后,打开连通阀,将分流物经管线由连通阀的第6接口进入,流到第1接口,经管线进入定量环收集。切换连通阀,打开第二维色谱,通过第二泵由第3接口经第4接口反冲定量环,使定量环收集的待测物由第1接口经第2接口进入第二分离柱进行分离,再经管线进入质谱进行测定。测定后,切换连通阀,使定量环内的多余待测物由第4接口至第5接口经管线排出。
实施例2
1、样品前处理
取2.000g的烟草粉末样品于50ml离心管中,加入10ml纯水振荡至样品被水充分浸润后静置10min。再加入内标溶液,置于涡旋振荡仪以2000r/min振荡1.0min。内标溶液为含内标样品的乙腈溶液,内标溶液中内标样品的浓度均为100ng/ml。
再加入quechers(quick、easy、cheap、effective、rugged、safe)试剂盒粉包,置于涡旋振荡仪以2000r/min振荡2.0min,再以5000r/min的速度离心10min,取第一上清液。试剂盒粉包内包括有4g无水硫酸镁、1g氯化钠和0.5g柠檬酸氢钠。
将1ml第一上清液放入quechers试剂盒含吸附剂小管中,用涡旋振荡仪以2000r/min振荡2.0min,再以5000r/min的速度离心5min,取第二上清液用0.22μm的有机相滤膜过滤后,即得待测样品。吸附剂小管内含150mg无水硫酸镁和25mgn-丙基乙二胺键合固定相吸附剂。
2、标准溶液的配制
分别取烟草中农残成分的标样,加入内标样品,再加入乙腈定容,配成混合标准溶液,混合标准溶液中农残成分的浓度均为10~500ng/ml,内标样品为三磷酸苯酯,内标样品的浓度为100ng/ml。
3、样品含量的测定
分别将2配制的标准样品和经样品前处理后待测样品进行中心切割二维色谱质谱联用系统检测,通过第一维色谱进行馏分后,馏分目标采用分流器分流、定量环收集,再由第二维色谱进行分离,通过质谱进行测定,比较保留时间、质谱扫描进行定性,确定待测样品中农残成分;同时采用内标标准曲线法进行mrm定量,获得待测样品中农残成分的含量。
具体来说,内标标准曲线法是先将上述2中一系列不同浓度的混合标准溶液分别进行中心切割二维色谱质谱联用系统检测,分别获得多种农残成分/相应内标物的二级选择离子峰面积比与多种农残成分/相应内标物的质量浓度比的线性关系,绘制相应的标准工作曲线,以多种农残成分与相应内标物的二级选择离子峰面积比为纵坐标(y轴),其多种农残成分与相应内标物的质量浓度比为横坐标(x轴),分别计算得到多种农残成分标准工作曲线的回归方程。
再将上述1中经样品前处理后待测样品溶液,进行中心切割二维色谱质谱联用系统检测,将获得的待测液中多种农残成分/相应内标物的二级选择离子峰面积比,代入步骤ⅰ中相应的多种农残成分标准工作曲线的回归方程,并根据加入相应内标物的已知质量浓度,计算得到待测样品溶液中多种农残成分的质量浓度。
其中,第一维色谱为体积排阻液相色谱(gpc)。第一维色谱的条件为:第一分离柱为tosohtsk-gelsupersw3000型凝胶色谱柱;紫外检测器的波长为230nm;柱温:30℃;流速:0.4ml/min;进样量:3μl;第一维流动相:40%乙腈。
分流器的分流比为7:1。馏分目标在定量环收集后要采用中心切割法,获得农残成分的馏分。即将第一维色谱分离出的目标物馏分的色谱峰的切割完整转移至第二维色谱,其它非目标物馏分不进第二维色谱。中心切割法的切割时间为8-14min。
第二维色谱为反相液相色谱(rplc)。第二维色谱的条件为:第二分离柱为thermoscientificacclaimpolaradvantageⅱc18(4.6*50mm,3μm)色谱柱;柱温:30℃;流速:0.4ml/min;进样量:3μl;第二维流动相a相:2mmol/l甲酸铵、0.1%甲酸、95%h2o+5%ch3cn(v:v);第二维流动相b相:2mmol/l甲酸铵、0.1%甲酸、5%h2o+95%ch3cn(v:v);梯度洗脱。
梯度洗脱的具体程序为:
0-17min,a相:b相体积比为95:5-95:5;
17-20min,a相:b相体积比为95:5-60:40;
20-44min,a相:b相体积比为60:40-20:80;
44-46min,a相:b相体积比为20:80-0:100;
46-56min,a相:b相体积比为0:100-0:100;
56-57min,a相:b相体积比为0:100-95:5;
57-60min,a相:b相体积比为95:5-95:5。
质谱的条件为:
质谱:agilenttechnologies6460triplequadms三重四极杆质谱;电离方式:电喷雾离子源(esi);扫描方式:正、负离子扫描,多反应监测(mrm);喷雾(is)电压为:5500v;雾化气(gas1)压力为:414kpa;气帘气(curtaingas)压力为:206.7kpa;辅助雾化气(gas2)压力为:482.6kpa;离子源温度(tem)为:500℃。
另外,本发明的中心切割二维色谱质谱联用系统在具体测定时,如图1所示,第一维色谱包括有依次连接的第一泵、第一分离柱、紫外检测器。第一泵与第一分离柱之间设有进样阀。
如图1所示,所述进样阀为二位六通阀,所述进样阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第4接口为进样口,所述第3接口为排废口,所述第1接口经管线与第一泵相连通,所述第6接口经管线与第一分离柱相连通。
如图1所示,所述第一泵为常规使用的液相色谱仪的专用泵。所述第一泵为四元泵。所述紫外检测器为常规使用的液相色谱仪的紫外检测器。
如图1所示,所述分流器为常规使用的分流器。所述定量环为常规使用的定量环(loop环)。所述定量环的容积为500μl;所述定量环的内径为1.00mm。
如图1所示,第二维色谱包括有依次连接的第二泵、第二分离柱。第二泵与第二分离柱之间设有连通阀。
如图1所示,所述连通阀为二位六通阀,所述连通阀设有第1接口、第2接口、第3接口、第4接口、第5接口、第6接口,所述第1接口和第4接口分别经管线与定量环的两端相连通,所述第2接口经管线与第二分离柱相连通,所述第3接口经管线与第二泵相连通,所述第5接口为排废口,所述第6接口经管线与分流器相连通。
如图1所示,所述第二泵为常规使用的液相色谱仪的专用泵。所述第二泵为四元泵。
如图1所示,采用中心切割二维色谱质谱联用系统进行测定时,打开第一维色谱和进样阀,第一维流动相通过第一泵由进样阀的第1接口进入,同时待测样品溶液由进样阀的第4接口进入,经第5接口,与第一维流动相由第6接口经管线进入第一分离柱进行馏分,并经紫外检测器检测。多余的第一维流动相,由进样阀的第1接口切换经第2接口至第3接口经管线排出。将馏分目标物经分流器分流后,打开连通阀,将分流物经管线由连通阀的第6接口进入,流到第1接口,经管线进入定量环收集。切换连通阀,打开第二维色谱,通过第二泵由第3接口经第4接口反冲定量环,使定量环收集的待测物由第1接口经第2接口进入第二分离柱进行分离,再经管线进入质谱进行测定。测定后,切换连通阀,使定量环内的多余待测物由第4接口至第5接口经管线排出。
实施例3
如上述实施例2中2所示,分别准确移取74种农残成分的标样,加入内标样品,再入加入乙腈定容,配成一系列混合标准溶液,其中内标样品的浓度为100ng/ml。
将上述配制好的一系列不同浓度的混合标准溶液分别进行中心切割二维色谱质谱联用系统检测,以74种农残成分与相应内标物的二级选择离子峰面积比为纵坐标(y轴),其74种农残成分与相应内标物的质量浓度比为横坐标(x轴),进行回归分析,得到回归方程的线性关系良好,在10-500ng/ml的线性浓度范围内,74种农残成分的回归方程的相关系数r均大于0.99,拥有良好的动态线性范围,具体数据见表1。
对混合标准溶液中的目标物响应信号,采用最低浓度标样重复进样10次,进行中心切割二维色谱质谱联用系统分析,以3倍标准偏差为方法的检出限(lod),10倍标准偏差为定量限(loq),不同农残成分的检出限(lod)和定量限(loq)见表1,具有较高的灵敏度。
表1方法线性相关系数及检出限、定量限
实施例4
取相同烟草样品考察方法的回收率、日内重复性、日间重复性,即选取已知农残成分浓度样品,加入已知浓度的混合标准溶液,然后进行上述实施例2中1的前处理,并进行中心切割二维色谱质谱联用系统分析,计算其回收率。同时对同一待测样品溶液分别在1日内、日间进行平行测定5次(n=5),得到74种农残成分的精密度测定数据,结果见表2。
由表2可以看出,目标物方法的回收率在72~136%之间,日内重复性的相对标准偏差(rsd)在1.6~10.2%之间,日间重复性的相对标准偏差(rsd)在3.3~13.6%之间,说明本发明方法的回收率高、准确性和重复性好,满足农残检测的需要。
实施例5
采用中心切割二维色谱质谱联用法与传统一维lc-ms/ms的方法进行比较,分别选取烟草样品。第一组采用传统一维lc-ms/ms的方法,参考标准yc/t184-2004,具体结果见图3。第二组采用上述实施例2中1的前处理,并进行中心切割二维色谱质谱联用系统分析,具体结果见图4。
在测定过程中每10个样品增加一个监控标样qc。从图3中可以发现做了20个样品之后质谱mrm总离子流图明显降低,从提取离子图中可以发现不同农残的质谱信号均有不同程度的降低。而如图4所示,本方法测定烟草农残100个样品后,监控标样信号无明显变化,说明本方法测定烟草中大批量农残可有效避免大分子基质影响,稳定性更突出。
所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!