一种泊沙康唑的高效液相色谱分析方法与流程
本发明涉及药物分析技术领域,具体而言,涉及一种泊沙康唑的高效液相色谱分析方法。
背景技术:
泊沙康唑(posaconazole,商品名诺科飞、noxafil)是新的吡咯类抗真菌药,由原先灵葆雅公司研发,其口服混悬液2005年11月在欧洲首次上市,美国fda2006年9月i5日批准上市,我国2013年6月通过cfda批准。泊沙康唑通过抑制麦角固醇的生物合成而发挥其抗真菌活性,属脂溶性药物,高脂饮食增加其吸收。临床用于治疗和预防侵袭性真菌感染,具有高效、低毒和广谱的特点,尤其对多烯类和其他三唑类药物的耐药菌,包括对隐球菌属、新型细球菌属、念珠菌属等感染均具有较好的临床疗效。
为了保证泊沙康唑后续的研发和生产质量,需要对原料药及其制剂的质量进行控制。因此,研究获得一种泊沙康唑的有关物质杂质检查和含量测定的检测方法,这对医药生产企业来说显得尤为迫切。在现有技术中,通常采用等度解决含量问题,采用梯度解决杂质问题,但因梯度的急剧变化常出现实验结果稳定性较差,数据偏差较大。
有鉴于此,有必要对现有技术中的泊沙康唑的含量及其杂质含量的分析方法予以改进,以解决上述问题。
技术实现要素:
本发明的目的在于提供一种泊沙康唑的高效液相色谱分析方法,它能够同时准确测定出泊沙康唑的含量和杂质,可有效地控制该产品的质量。
为实现上述发明目的,本发明提供了一种泊沙康唑的高效液相色谱分析方法:将含有泊沙康唑的样品溶液通入色谱柱中,采用反相液相色谱外标百分比法定量分析,所述液相色谱条件为:
色谱柱:采用十八烷基硅烷键合硅胶为填充材料,规格4.6*250mm,3μm;
流动相:体积配比为50:50-60:40的流动相a与流动相b的混合溶液,等度洗脱;
检测波长为205nm~220nm;
流速为1.0~2.0ml/min;
柱温为25~35℃;
进样体积为10~30μl。
在一些实施方式中,流动相a中磷酸、四甲基氢氧化铵、水的质量配比为(2~10):(5~20):(1000~2000),所述流动相b中水和乙腈的体积配比为(50~200):(800~950)。
在一些实施方式中,样品溶液由含有泊沙康唑的制剂与稀释剂组成。
在一些实施方式中,稀释剂由(0.1%~0.3%)三乙胺与甲醇组成,所述(0.1%~0.3%)三乙胺溶液与甲醇的体积配比为20:80~30:70。
在一些实施方式中,稀释剂中0.1%三乙胺溶液与甲醇的体积配比为25:75。
在一些实施方式中,含有泊沙康唑的制剂为泊沙康唑原料、泊沙康唑片剂、泊沙康唑注射剂或者泊沙康唑口服混悬剂。优选为泊沙康唑片剂。
在一些实施方式中,检测波长为215nm,流速为1.7ml/min,柱温为30℃,进样体积为20μl。
与现有技术相比,本发明的有益效果是:本发明所提供的泊沙康唑的高效液相分析方法具有实用可靠、稳定性较好、数据重现性强的优点,并且能够从泊沙康唑中有效分离出2个关键杂质,即杂质rrt0.88、与杂质rrt0.93,可有效地控制该产品的质量。
附图说明
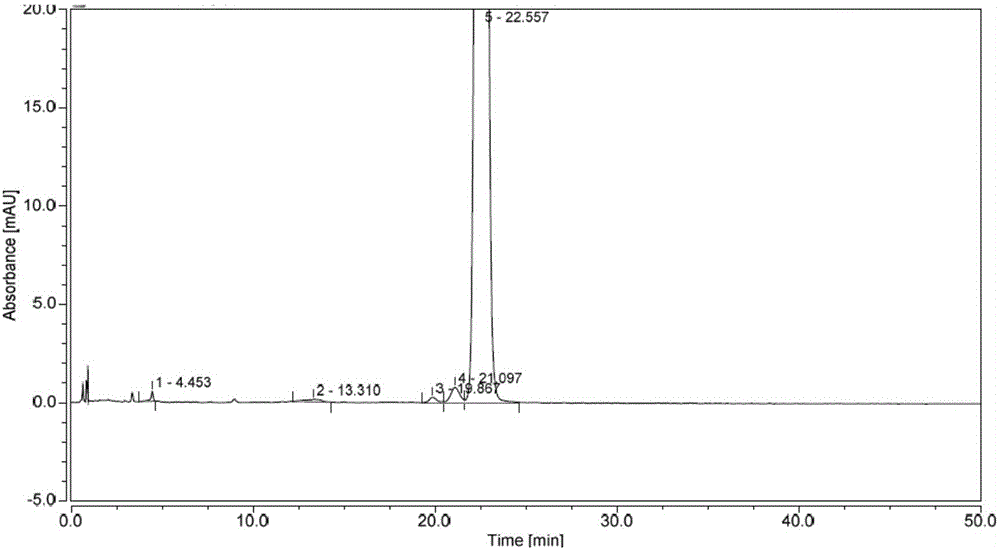
图1是本发明实施例一中的泊沙康唑原料的高效液相色谱图;
图2是本发明实施例二中的泊沙康唑制剂的高效液相色谱图;
图3是本发明实施例一中的泊沙康唑空白辅料的高效液相色谱图。
具体实施方式
下面结合附图所示的各实施方式对本发明进行详细说明,但应当说明的是,这些实施方式并非对本发明的限制,本领域普通技术人员根据这些实施方式所作的功能、方法、或者结构上的等效变换或替代,均属于本发明的保护范围之内。
实施例一:
本实施例提供了一种泊沙康唑的高效液相色谱分析方法,针对泊沙康唑原料含量和杂质的测定。液相色谱条件为:
色谱柱:ymcpackc18,规格为4.6*250mm,3μm
流动相:流动相a:流动相b=55:45(体积配比),等度洗脱,流动相a中磷酸、四甲基氢氧化铵、水的质量配比为6:9:2000,流动相b中水、乙腈的体积配比为150:850。
检测波长为215nm;
流速为1.7ml/min;
柱温30℃;
进样体积20μl;
稀释剂:0.1%三乙胺:甲醇=25:75(体积配比)。
实验步骤:
溶液配制:
取泊沙康唑原料约50mg,精密称定,置于100ml容量瓶中,加适量稀释剂溶解并稀释至刻度,摇匀,作为供试品溶液。
精密量取供试品溶液1ml,置100ml量瓶中,加稀释剂稀释至刻度,摇匀,作为杂质对照溶液。
精密称取泊沙康唑对照品50mg,置100ml量瓶中,加稀释剂至刻度,摇匀,作为含量对照品溶液。
按照上述分析方法精密量取供试品溶液、杂质对照溶液和含量对照品溶液各20μl注入液相色谱仪,记录液相色谱图,如图1所示。按照不加校正因子的自身对照法计算其杂质,按照外标法计算其含量。
结果表明,空白溶剂不干扰泊沙康唑原料的含量检测,也不干扰其杂质检测。泊沙康唑原料中各杂质可以得到有效分离,分离度均大于1.5,峰形良好,峰纯度良好,理论塔板数大于3000,拖尾因子小于1.5,符合标准。
本发明提供的分析方法,根据中国药典2015版,完成方法学验证,包括系统适性试验、破坏性试验、影响因素试验、回收率试验、以及耐用性试验。经过这些验证,本发明提供的分析方法实用可靠,稳定性较好。
实施例二:
本实施例提供了一种泊沙康唑的高效液相色谱分析方法,针对泊沙康唑缓释片含量和杂质的测定。液相色谱条件为:
色谱柱:ymcpackc18,规格为4.6*250mm,3μm。
流动相配比为:流动相a:流动相b=55:45(体积配比),等度洗脱,流动相a中磷酸、四甲基氢氧化铵、水的质量配比为6:9:2000,流动相b中水、乙腈的体积配比为150:850;
检测波长为215nm;
流速为1.7ml/min;
柱温30℃;
进样体积20μl;
稀释剂:0.1%三乙胺:甲醇=25:75(体积配比)。
实验步骤:
溶液配制:
取泊沙康唑片剂适量,研细,精密称定(相当于泊沙康唑50mg),置于100ml容量瓶中,加适量稀释剂,45℃超声30分钟、稀释剂定容至刻度,摇匀,3000rpm离心5分钟,取上清液用0.45微米滤膜过滤,续滤液作为供试品溶液。
精密量取供试品溶液1ml,置100ml量瓶中,加稀释剂稀释至刻度,摇匀,作为杂质对照溶液;取片剂空白辅料适量,置于1000ml容量瓶中,加适量稀释剂,45℃超声30分钟、稀释剂定容至刻度,摇匀,3000rpm离心5分钟、取上清液用0.45微米滤膜过滤,续滤液作为空白辅料溶液。
精密称取泊沙康唑对照品50mg,置100ml量瓶中,加稀释剂至刻度,摇匀,作为含量对照品溶液。
精密量取供试品溶液、杂质对照溶液和含量对照品溶液各20μl注入液相色谱仪,记录液相色谱图,如图2和图3所示。按照不加校正因子的自身对照法计算其杂质,按照外标法计算其含量。
结果表明,空白辅料溶液不干扰泊沙康唑缓释片的含量检测,也不干扰其杂质检测。泊沙康唑缓释片中各杂质可以得到有效分离,分离度均大于1.5,峰形良好,峰纯度良好,理论塔板数大于3000,拖尾因子小于1.5,符合标准。
泊沙康唑缓释片含量结果:相当于标示量的99.2%;杂质结果:杂质rrt0.88的含量为0.14%,杂质rrt0.93的含量为0.33%。泊沙康唑缓释片中关键的杂质均能准确测定出。同时对泊沙康唑缓释片进行破坏试验,降解杂质也得到了有效的分离,分离度大于1.5,主峰纯度较好,本发明可以有效的评价泊沙康唑缓释片的质量。
本发明提供的分析方法,根据中国药典2015版,完成方法学验证,包括系统适性试验、破坏性试验、影响因素试验、回收率试验、以及耐用性试验。经过这些验证,本发明提供的分析方法实用可靠,稳定性较好。
实施例三:
本实施例提供了一种泊沙康唑的高效液相色谱分析方法,针对泊沙康唑注射剂的含量和杂质的测定。液相色谱条件为:
色谱柱:ymcpackc18,规格为4.6*250mm,3μm。
流动相配比为:流动相a:流动相b=55:45(体积配比),等度洗脱,流动相a中磷酸、四甲基氢氧化铵、水的质量配比为6:9:2000,流动相b中水:乙腈的体积配比为150:850;
检测波长为215nm;
流速为1.7ml/min;
柱温30℃;
进样体积20μl;
稀释剂:0.1%三乙胺:甲醇=25:75(体积配比)。
实验步骤:
溶液配制:
取泊沙康唑注射剂适量(相当于泊沙康唑50mg),精密量取,置于100ml容量瓶中,加适量稀释剂,45℃超声5分钟、稀释剂定容至刻度,摇匀,3000rpm离心5分钟、取上清液用0.45微米滤膜过滤,续滤液作为供试品溶液。
精密量取供试品溶液1ml,置100ml量瓶中,加稀释剂稀释至刻度,摇匀,作为杂质对照溶液。
取注射剂空白辅料适量,置于100ml容量瓶中,加适量稀释剂,45℃超声5分钟、稀释剂定容至刻度,摇匀,3000rpm离心5分钟、用0.45微米滤膜过滤,续滤液作为空白辅料溶液。
精密称取泊沙康唑对照品50mg,置100ml量瓶中,加稀释剂至刻度,摇匀,作为含量对照品溶液。精密量取供试品溶液、杂质对照溶液和含量对照品溶液各20μl注入液相色谱仪,记录液相色谱图。按照不加校正因子的自身对照法计算其杂质,按照外标法计算其含量。
结果表明,空白辅料不干扰泊沙康唑注射剂的含量检测,也不干扰其杂质检测。泊沙康唑注射剂中各杂质可以得到有效分离,分离度均大于1.5,峰形良好,峰纯度良好,理论塔板数大于3000,拖尾因子小于1.5,符合标准。
实施例四:
本实施例提供了一种泊沙康唑的高效液相色谱分析方法,针对泊沙康唑口服混悬剂的含量和杂质的测定。液相色谱条件为:
色谱柱:ymcpackc18,规格为4.6*250mm,3μm。
流动相配比为:流动相a:流动相b=55:45(体积配比),等度洗脱,流动相a中磷酸、四甲基氢氧化铵、水的质量配比为6:9:2000,流动相b中水:乙腈的体积配比为150:850;
检测波长为215nm;
流速为1.7ml/min;
柱温30℃;
进样体积20μl;
稀释剂:0.1%三乙胺:甲醇=25:75(体积配比)。
实验步骤:
溶液配制:
取泊沙康唑口服混悬剂适量(相当于泊沙康唑50mg),精密量取,置于100ml容量瓶中,加适量稀释剂,45℃超声5分钟,稀释剂定容至刻度,摇匀。3000rpm离心5分钟,取上清液用0.45微米滤膜过滤,续滤液作为供试品溶液。
精密量取供试品溶液1ml,置100ml量瓶中,加稀释剂稀释至刻度,摇匀,作为杂质对照溶液。
取片剂空白辅料适量,置于100ml容量瓶中,加适量稀释剂,45℃超声5分钟、稀释剂定容至刻度,摇匀,3000rpm离心5分钟、用0.45微米滤膜过滤,续滤液作为空白辅料溶液。
精密称取泊沙康唑对照品50mg,置100ml量瓶中,加稀释剂至刻度,摇匀,作为含量对照品溶液。
精密量取供试品溶液、杂质对照溶液和含量对照品溶液各20μl注入液相色谱仪,记录液相色谱图。按照不加校正因子的自身对照法计算其杂质,按照外标法计算其含量。
结果表明,空白辅料溶液不干扰泊沙康唑口服混悬剂的含量检测,也不干扰其杂质检测。泊沙康唑口服混悬剂中各杂质可以得到有效分离,分离度均大于1.5,峰形良好,峰纯度良好,理论塔板数大于3000,拖尾因子小于1.5,符合标准。
上文所列出的一系列的详细说明仅仅是针对本发明的可行性实施方式的具体说明,它们并非用以限制本发明的保护范围,凡未脱离本发明技艺精神所作的等效实施方式或变更均应包含在本发明的保护范围之内。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
- 还没有人留言评论。精彩留言会获得点赞!