一种分析桂花中水溶性精油成分的方法与流程
本发明涉及一种分析桂花中水溶性精油成分的方法,具体涉及一种超声原位生成二氧化碳气体冒泡辅助分散液液微萃取技术结合气相色谱/质谱法分析桂花中水溶性精油成分(芳香水和浸渍水中的挥发性成分)的方法。
(二)
背景技术:
桂花,属于木犀科木犀属的植物,广泛分布在中国、日本、东南亚和一些欧洲国家。桂花在我国己有2500年的栽培历史,是我国十大传统名花之一。因有特殊的香味,早在东汉之年桂花就被誉为神仙之树,“吴刚伐桂”更是家喻户晓的神话故事,可见桂花在中国历史文化中起着重要的作用。目前,桂花作为景观植物在中国南方广泛种植。随着天然产物研究的深入,桂花具有药理活性,例如强抗氧化性,神经保护作用,降血糖以及抑制黑色素生成。桂花有很多品种,根据花的颜色主要有银桂、丹桂、金桂三个品种。文献报道显示,桂花没有花蜜,但依然吸引了大量昆虫完成授粉。由于甜美宜人的香味,桂花更是可作为食品的香料及甜食的添加物,例如糕点、蜜饯等;也可制作浸酒及配制药膳等。除此之外,桂花茶也是一种民间较喜欢的饮料。
植物中精油的萃取方法有许多,例如溶剂萃取、超临界流体萃取、同时蒸馏萃取、水蒸馏或蒸汽蒸馏。但当植物中精油含量较少时,这些方法需要较多的待萃取植物的样品量。水溶性精油是指精油含量较低时溶解在水溶液中的部分,其包括两种形式:1.植物芳香水,即蒸馏所收集的馏分;2.植物的浸渍水,其中茶饮水就属于植物浸渍水的一种形式。植物中水溶性精油因其含量较低,现有分析方法中较难直接萃取出其中的成分。因此,在分析过程中需要对的水样进行富集。
目前,植物中水溶性精油成分可以利用以下几种技术进行分析。磁性固相萃取是一种利用磁性吸附剂吸附待测物的方法,其最大的优点是在外加磁场下磁性吸附剂会与水体分离,为分离带来了便捷。大量新型吸附剂也已应用于不同残留物的检测分析,但磁性固相材料相对复杂的制备过程限制了该方法的应用。固相微萃取技术是集提取与浓缩于一步的一种无溶剂的样品前处理方法,该方法对挥发性成分的分析检测具有优势;对于一些难挥发的化合物的分析则需直接浸渍到样品中,但这会破坏萃取纤维而影响方法的稳定性。在固相微萃取技术基础上发展的搅拌棒吸附萃取技术应用于水样的萃取也有文献报道,但该方法操作时间过长。中空纤维膜液相微萃取和单滴微萃取也可对水样中的目标物进行分析。而上述两种方法属于液相微萃取技术,此项技术在过去的的二十年中吸引了大量研究者的关注,其最大的特点就是采用少量的萃取剂,获得较高的富集因子,具有较好的应用前景。
2006年,rezaee提出一种新的液相微萃取技术——分散液液微萃取技术,这项技术是基于水溶液与疏水的有机萃取溶剂在两亲性分散剂的协助下形成乳浊液,而后通过离心使乳浊液变澄清(即破乳),使得有机相与水溶液样品分离。分散液液微萃取技术在分析植物中的挥发性成分方面已有许多文献报道,雅雯等人开发了一种分散液液微萃取腊梅鲜花的挥发性成分的技术,将植物样品浸入有机溶剂进行超声提取,然后将提取液作为分散剂用到分散液液微萃取技术的分析中。sereshti等将植物样品直接浸入有机溶剂用超涡旋方式提取挥发性性组分,再引入分散液液微萃取技术进行萃取和富集。但上述这些方法都需要离心使水溶液与有机溶剂分离,一方面限制了大体积水溶液样品的应用,另一方面富集倍数较小。此外,分散剂的使用也会使得萃取效率降低。
本发明致力于建立一种超声原位生成二氧化碳气体冒泡辅助分散液液微萃取技术结合气相色谱/质谱法分析桂花中水溶性精油成分的方法。该方法利用超声冒泡辅助破乳技术,使得乳浊液迅速变澄清而不需额外的离心设备,具有富集倍数高、耗时短等特点,可对水中浓度较低的化合物进行富集,并在此基础上进行分析测定。
(三)
技术实现要素:
针对现有技术中存在的不足,本发明提供了一种超声原位生成二氧化碳气体冒泡辅助分散液液微萃取技术结合气相色谱/质谱法分析桂花中水溶性精油成分的方法,本发明方法具有富集倍数高、耗时短的特点,是一种能够快速高效萃取桂花中水溶性精油成分的新方法,并且有适用于提取其他植物水溶性精油成分的潜力。
本发明的基本构思充分利用了超声冒泡辅助破乳技术,使得乳浊液迅速变澄清而不需额外的离心设备的特点。本发明的方法所需设备简单,操作简便且操作时间短,可以适用于不同体积样品的前处理操作。同时该方法适用于对水中浓度较低的化合物进行富集,并在此基础上进行分析测定。
本发明的技术方案如下:
一种超声原位生成二氧化碳气体冒泡辅助分散液液微萃取技术结合气相色谱/质谱法分析桂花中水溶性精油成分(芳香水和浸渍水中的挥发性成分)的方法,所述方法包括如下步骤:
(1)新鲜桂花芳香水和浸渍水的制备
新鲜桂花芳香水的制备:将新鲜桂花与去离子水按料液比1:5混合,得到原料液,在微波功率为200w下进行水蒸气蒸馏,当蒸出的馏分体积为原料液体积的1/4~1/3时,停止蒸馏,取馏分用去离子水稀释3~4倍,得到桂花芳香水,4℃下保存待用;
新鲜桂花浸渍水的制备:将新鲜桂花与85~95℃的去离子水按料液比1:5混合,得到原料液,保温浸渍30min,过滤,取滤液用去离子水补足体积至与原料液体积相等,得到桂花浸渍水,4℃下保存待用;
所述新鲜桂花例如可以为下列中的至少一种:银桂(osmanthusfragransvar.aurantiacus)、丹桂(osmanthusfragransvar.aurantiacus)、金桂(osmanthusfragransvar.thunbergii.)、四季桂(osmanthusfragransvar.semperflorens);
(2)分散液液微萃取
取待测水样,用去离子水稀释后作为萃取样品,在萃取样品中加入质子供体、萃取溶剂预混合后,超声乳化,然后加入二氧化碳源,超声冒泡破乳,吸取上层萃取溶剂相,经无水硫酸钠干燥后,得到样品溶液;
所述待测水样为步骤(1)制备的新鲜桂花芳香水或浸渍水;
稀释用去离子水的体积量为待测水样体积的20倍;
所述质子供体为盐酸、乙酸、柠檬酸或磷酸二氢钠,优选磷酸二氢钠;
所述萃取溶剂为甲苯;
所述二氧化碳源为碳酸钠或碳酸氢钠,推荐二氧化碳源以0.44~1.75mol/l水溶液的形式投加;
所述质子供体与二氧化碳源的物质的量之比为1~6:1,优选4:1;
所述二氧化碳源的物质的量以待测水样的体积计为8~10mol/l,优选8.8mol/l;
所述萃取溶剂与待测水样的体积比为5~7:100,优选5.5:100;
所述超声的功率在250w,所述超声乳化的时间为15~90s(优选30s);所述超声冒泡破乳的时间为60~180s(优选90s);
(3)样品检测
将步骤(2)所得样品溶液注入气相色谱-质谱联用仪进行分析,得到样品气相色谱质谱总离子流图,通过与标准物质谱图进行对照,得出样品溶液中所含成分;
气相色谱条件:色谱柱为db-5ms(长度30m×内径0.25mm×膜厚0.25μm);进样口温度:250℃,不分流进样;柱温在40℃保持3min,然后以5℃·min-1升到90℃,保持1min,最后以3℃·min-1升到250℃,保持5min;载气为高纯氦气(99.999%),流速为0.8ml·min-1,进样量1μl;
质谱条件:电子轰击离子源(ei),电子能量为70ev;采用全扫描模式,扫描范围在45~550m/z;离子阱温度为180℃,传输线温度为250℃,箱体温度为50℃;扫描速度为3scans·s-1,溶剂延迟为5min。
与现有技术相比,本发明的有益效果为:
1、本发明提供了一种新型、有效分析桂花中水溶性精油成分的方法;
2、首次采用超声原位生成二氧化碳气体冒泡辅助破乳与分散液液微萃取技术结合,克服了传统分散液液微萃取方法中使用离心进行破乳的问题,具有富集倍数高、耗时短等特点;
3、采用超声冒泡辅助破乳技术,使得乳浊液迅速变澄清而不需额外的离心设备;
4、本发明所需设备简单,操作简便且操作时间短,可以适用于不同体积样品的前处理操作;
5、应用本发明能结合实际,是一种能够快速高效萃取桂花中水溶性精油成分的新方法,并且为提取其他植物水溶性精油成分提供一个新的手段。
(四)附图说明
图1为本发明萃取桂花中水溶性精油成分的装置图;
图2a、2b、2c、2d、2e、2f、2g分别为实施例1中的酸(质子供体)的种类、酸(质子供体)与碱(二氧化碳源)的比例、生成二氧化碳的气体量、碳酸钠浓度、萃取剂体积、超声乳化时间、冒泡破乳时间的优化结果;
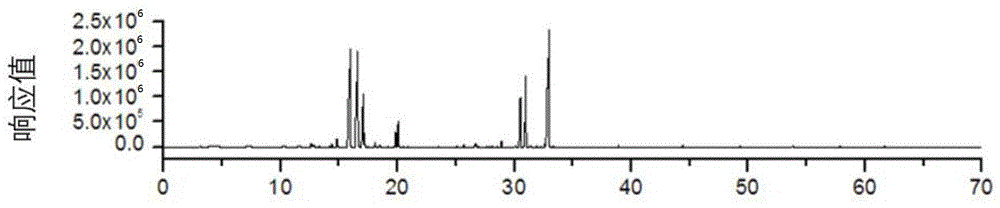
图3为实施例1中银桂的芳香水挥发性成分总离子流图;
图4为实施例1中银桂的浸渍水挥发性成分总离子流图;
图5为实施例1中银桂花中的挥发性成分总离子流图。
(五)具体实施方式
下面通过具体实施例对本发明作进一步描述,但本发明的保护范围并不仅限于此。
实施例1:银桂水溶性精油成分的分析方法
(1)银桂芳香水和浸渍水的制备
新鲜银桂芳香水制备:其通过微波辅助水蒸气蒸馏制备而得的馏分。在常压微波装置依次设置微波参数如下:功率为200w,初始时间为1h。将20.0g银桂桂花与100ml去离子水置于反应烧瓶(250ml)中,然后将烧瓶置于微波反应室,通过微波仪器顶部的孔将其与蒸馏装置相连。当芳香水为30ml时停止蒸馏,将其取出并定容至100ml,在4℃下保存用于后续实验。
新鲜银桂浸渍水制备:将20.0g新鲜桂花样品浸入100ml热水(90±2℃)中;30分钟后取出滤液,将其取出并定容至100ml,并在4℃下保存用于后续实验。
(2)新鲜银桂样品芳香水和浸渍水的分散液液微萃取步骤
移取1ml芳香水或浸渍水到自制分散液液微萃取装置(图1),之后用去离子水稀释至20ml作为萃取样品。首先,将4.20g磷酸二氢钠粉末放入样品中振荡溶解;然后,向瓶中加入55μl甲苯作为萃取剂,将溶液预混合后,将烧瓶置于超声浴中乳化30s;接下来,向侧管中加入10ml的碳酸钠(0.88mol·l-1),之后放入超声浴中超声加速二氧化碳(8.75×10-3mol)冒泡破乳90s。超声结束后,向侧管中加入饱和盐水以提高液面毛细管的位置,用气相进样针将上层甲苯相取出,并转移到装有无水硫酸钠的离心管中。最后,取经干燥处理的甲苯有机相1μl注入气相色谱-质谱联用仪的进样口。
(3)样品检测
将经过前处理的样品注入气相色谱-质谱联用仪进行分析,得到样品气相色谱质谱总离子流图;
气相色谱条件:色谱柱为db-5ms(长度30m×内径0.25mm×膜厚0.25μm);进样口温度:250℃,不分流进样;柱温在40℃保留3min然后以5℃·min-1升到90℃,在此温度下保持1min,最后以3℃·min-1升到250℃后保持5min;载气为高纯氦气(99.999%),流速为0.8ml·min-1。
质谱条件:电子轰击离子源(ei),电子能量为70ev;采用全扫描模式,扫描范围在45-550m/z;离子阱温度为180℃,传输线温度为250℃,箱体温度为50℃;扫描速度为3scans·s-1,溶剂延迟为5min。
成分定性通过采集所得到的质谱图利用nist(2011版本)谱库检索完成;同时采用kovats保留指数定性的方法来辅助质谱检索定性。研究证实,质谱检索与保留指数相结合的二维定性是一种可信度较高的定性方法。在进行保留指数定性时,参考色谱柱选用db-5ms或db-5的文献值,而实验值与文献值的差异一般以1%作为检索尺度。研究中所用的正构烷烃标准样品为c8-c30,利用峰面积归一化方法进行相对含量的定量分析。
(4)方法评估
使用相对标准偏差评估方法的精确度,在最优条件下,重复测量银桂芳香水样品6次。5种主要化合物(芳樟醇氧化物b,芳樟醇氧化物a,α-紫罗兰酮,二氢-β-紫罗兰酮,β-紫罗兰酮)的峰面积的rsd值分别为7.8%,10.7%,8.0%,11.2%和7.8%,总峰面积的rsd值为9.2%。结果表明,该方法对挥发性成分的萃取具有良好的精密度,适用于对挥发性成分的分析。
为了评估方法,与其他方法进行对比,其实验条件如下:
固相微萃取法:将银桂鲜花样品(0.13±0.01g)放入20ml顶空玻璃萃取瓶中,室温30℃±2℃下平衡30分钟;然后用dvb/car/pdms(50/30μm,2cm)的spme萃取杆通过顶空瓶的密封垫片插入萃取瓶中,并将其纤维暴露于样品上方的顶部空间30分钟;萃取完成后,将spme萃取杆拔出并直接插入气相色谱-质谱联用仪的进样口。
通过质谱图利用nist(2011版本)谱库检索和kovats保留指数定性的方法进行成分定性,比较采用分散液液微萃取水溶性精油(芳香水和浸渍水)和固相微萃取三个谱图,分别鉴定出47种,39种,和29种的挥发性组分。各组分的相对含量是通过峰面积归一化方法计算所得。在银桂芳香水中主要成分是β-紫罗兰酮(38.92%)、二氢-β-紫罗兰酮(11.68%)、芳樟醇氧化物a(8.34%)、芳樟醇氧化物b(6.93%)和α-紫罗兰酮(5.33%)。银桂茶饮水中主要成分是β-紫罗兰酮(37.28%)、芳樟醇(18.76%)和联苄(5.58%)。采用固相微萃取银桂自然花香中的主要成分为β-紫罗兰酮(26.38%)、芳樟醇氧化物b(20.20%)、芳樟醇氧化物反式(20.11%)、二氢β-紫罗兰酮(8.51%)和α-紫罗兰酮(6.06%)。分析表明,在自然花香中,低沸点化合物占很大比例。通过挥发性成分总离子流图比较,采用分散液液微萃取测定得到的水溶性精油(芳香水和浸渍水)中挥发性成分比采用固相微萃取测定新鲜银桂样品中的挥发性成分更为复杂;采用相同量的桂花鲜花样品所制得的浸渍水与芳香水,在相同的条件下进行分散液液微萃取对其挥发性成分进行萃取和富集,芳香水中挥发性成分性组分的绝对含量比浸渍水多。总体来说,该方法作为是一种快速、简单、环保、可靠的分析桂花中水溶性精油成分的新方法,有适用于提取其他植物挥发性成分的潜力。
- 还没有人留言评论。精彩留言会获得点赞!