一种检测王不留行药材、饮片、标准汤剂、配方颗粒特征图谱的高效液相方法与流程
本发明具体涉及一种王不留行药材、饮片、标准汤剂、配方颗粒特征图谱的hplc检测方法。
背景技术:
王不留行为石竹科植物麦蓝菜vaccarriaseetalis(neck.)garcke的干燥成熟种子,有活血通经,下乳消肿,利尿通淋之功,可治经闭,痛经,乳汁不下,乳痈肿痛,淋症涩痛等病症,疗效显著,为临床常用中药品种。王不留行中主要含有三萜皂苷、环肽、黄酮、生物碱、酚酸、类固醇和挥发油等成分。临床研究表明,王不留行能治疗乳汁分泌不足,慢性前列腺炎,乳腺增生症,急性腰扭伤,突发性耳聋,外用治疗带状疱症,桡骨茎突下载性腱鞘炎等,这些化学成分的整体组成与含量是王不留行发挥临床疗效的基础,因此,要确保其临床疗效,需要对其药效成分进行整体质量控制。
目前,《中国药典》2015年版一部王不留行项下仅收载采用高效液相色谱法测定王不留行黄酮苷单一成分含量的方法,尚未进行其特征图谱控制。其他有部分文献研究报道建立了王不留行饮片及其炮制前后的成分及含量变化,如周国洪.王不留行化学成分及炮制对其影响研究[d].中国中医科学院,2016,5;陈琳,胡昌江,冯建,等.炒王不留行配方颗粒高效液相指纹图谱研究[j].亚太传统医药,2013,9(4):16-18;黄懿,欧阳波,肖作奇,等.不同产地炒王不留行hplc指纹图谱及含量测定研究[j].中南药学,2017(7):879-882.
然而,上述报道仅针对王不留行饮片及炮制品研究建立的控制指标,检测方法各不相同,未见标准汤剂、配方颗粒指纹图谱研究的报道。因此,现有的检测方法难以有效对比和分析王不留行药材、饮片、标准汤剂、配方颗粒特征图谱的差异与变化,难以深入认识王不留行药效物质基础在从原料到配方颗粒成品的工艺过程的传递情况,难以整体评价和控制从王不留行药材到配方颗粒成品的工艺过程。因此,建立统一的王不留行药材、饮片、标准汤剂、配方颗粒特征图谱测定方法,有利于整体评价王不留行相关工艺过程的科学性、合理性,更能整体控制王不留行药材、饮片、标准汤剂、配方颗粒的内在质量,确保王不留行的临床疗效。
技术实现要素:
为解决上述问题,本发明提供了一种适用于王不留行药材、饮片、标准汤剂、配方颗粒特征图谱测定的hplc检测方法。
本发明的整体控制王不留行药材、饮片、标准汤剂、配方颗粒的高效液相特征图谱检测方法,包括如下操作步骤:
1)对照品溶液制备:取对照品,溶解,即得对照品溶液;
2)对照药材溶液制备:取王不留行对照药材,经提取,即得对照药材溶液;
3)供试品溶液制备:取王不留行药材、饮片、标准汤剂、配方颗粒,经提取,即得供试品溶液;
4)分别吸取对照品溶液、对照药材溶液和供试品溶液注入高效液相色谱仪,色谱条件如下:
色谱柱:以十八烷基硅烷键合硅胶为填充剂;检测波长:240~270nm;流动相:乙腈为流动相a,以0.1%磷酸溶液为流动相b,梯度洗脱程序如下:
其中,步骤1)所述对照品包括:王不留行黄酮苷、原儿茶酸、刺桐碱、肥皂草苷、王不留行环肽a、王不留行环肽b、异牡荆素-2”-o-阿拉伯糖苷七种对照品。
其中,步骤1)所述溶解是加入甲醇或70%甲醇溶液溶解。
其中,步骤2)所述提取是指:加入70%甲醇溶液,超声提取。
其中,步骤3)所述提取是指:加入70%甲醇溶液,超声提取。
优选地,所述提取方法如下:
取王不留行药材或饮片,加入10倍70%甲醇,或取王不留行标准汤剂粉末,加入100倍70%甲醇,或取王不留行配方颗粒研细后粉末,加入50倍70%甲醇;超声提取30分钟,放冷,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得王不留行标准汤剂或配方颗粒的供试品溶液;所述超声提取的功率为600w、频率为40khz。
其中,所用色谱柱为agilentzorbaxeclipsexdb-c18色谱柱,规格均为4.6mm×250mm,5μm。
其中,所述检测波长为270nm。
其中,所述色谱条件还包括:流速为1.0ml/min,柱温为30℃,进样量为10μl。
本发明的王不留行药材、饮片、标准汤剂、配方颗粒高效液相特征图谱的检测方法,能整体控制王不留行药材、饮片、标准汤剂、配方颗粒中的特征成分,确保王不留行药材、饮片、标准汤剂、配方颗粒整体质量的稳定,且方法操作简单,精密度高,稳定性好,重复性好,准确度高。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
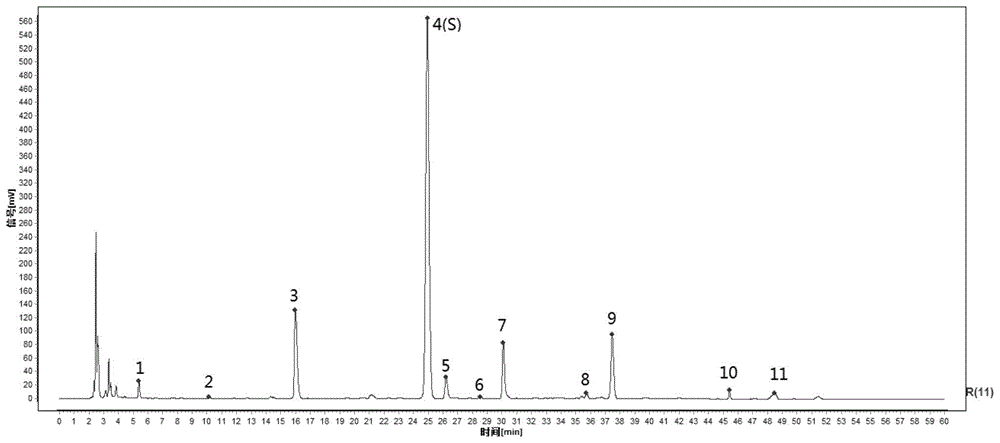
图1王不留行药材对照图谱,峰2:原儿茶酸;峰3:刺桐碱;峰4(s):王不留行黄酮苷;峰5:肥皂草苷;峰7:异牡荆素-2”-o-阿拉伯糖苷;峰10:王不留行环肽b;峰11:王不留行环肽a。
图2王不留行饮片对照图谱,峰2:原儿茶酸;峰3:刺桐碱;峰4(s):王不留行黄酮苷;峰5:肥皂草苷;峰7:异牡荆素-2”-o-阿拉伯糖苷;峰10:王不留行环肽b;峰11:王不留行环肽a。
图3王不留行标准汤剂对照图谱,峰2:原儿茶酸;峰3:刺桐碱;峰4(s):王不留行黄酮苷;峰5:肥皂草苷;峰7:异牡荆素-2”-o-阿拉伯糖苷;峰10:王不留行环肽b;峰11:王不留行环肽a。
图4王不留行配方颗粒对照图谱,峰2:原儿茶酸;峰3:刺桐碱;峰4(s):王不留行黄酮苷;峰5:肥皂草苷;峰7:异牡荆素-2”-o-阿拉伯糖苷;峰10:王不留行环肽b;峰11:王不留行环肽a。
图5药材、饮片、标准汤剂、配方颗粒对照特征图谱叠加图,峰2:原儿茶酸;峰3:刺桐碱;峰4(s):王不留行黄酮苷;峰5:肥皂草苷;峰7:异牡荆素-2”-o-阿拉伯糖苷;峰10:王不留行环肽b;峰11:王不留行环肽a。
具体实施方式
仪器与试药
1仪器
高效液相色谱仪:安捷伦1260型高效液相色谱仪、waterse2695型高效液相色谱仪、岛津30ad型高效液相色谱仪;
电子天平:me204e/02、ms205du、xp26(梅特勒-托利多仪器有限公司);
超纯水机:细胞型1810a(上海摩勒科学仪器有限公司);
超声波清洗器:kq-600db型(600w,40khz;昆山市超声仪器有限公司);
色谱柱:zorbaxeclipsexdb-c18analytical4.6×250mm5-micron、diamonsil5μmc18(2)250×4.6mm、shim-packgist5μmc184.6×250mm。
2试药
乙腈、磷酸为色谱纯,水为超纯水,其余试剂均为分析纯。
王不留行黄酮苷(中国食品药品检定研究院,批号:111853-201704,含量以96.9%计);原儿茶酸(中国食品药品检定研究院,批号:110809-201205,含量以99.9%计);刺桐碱(四川省维克奇生物科技有限公司,批号:wkq18012302,含量≥98%);肥皂草苷(四川省维克奇生物科技有限公司,批号:wkq18011711);王不留行环肽a(成都曼斯特生物科技有限公司,批号:must-18033102,含量以99.24%计);王不留行环肽b(成都曼斯特生物科技有限公司,批号:must-18051007,含量以99.6%计);异牡荆素-2”-o-阿拉伯糖苷(四川省维克奇生物科技有限公司,批号:wkq18010308,含量≥98%);王不留行对照药材(中国食品药品检定研究院,批号:121094-201305);
王不留行配方颗粒(四川新绿色药业科技发展有限公司制备,批号:1804009、sy1807001、sy1807002、sy1807003)。
王不留行药材(批号:xls201805338、xls201805339、xls201805340、xls201805341、xls201805343、xls201805344、010458-1706001、xls201806513、xls201806514、xls201806515、xls201806516、xls201806517、xls201806563、xls201806564、xls201806565、xls201806566、xls201806567、xls201806568、xls201806569、xls20180670、xls201806571、xls201806572);
王不留行饮片(批号:wblx180701、wblx180702、wblx180703、wblx180704、wblx180705、wblx180706、wblx180707、wblx180708、wblx180709、wblx180710、wblx180711、wblx180712、wblx180713、wblx180714、wblx180715、wblx180716、wblx180717、wblx180718、wblx180719、wblx180720、wblx180721、wblx180722);
王不留行标准汤剂(四川新绿色药业科技发展有限公司制备,批号:wblxbt180701、wblxbt180702、wblxbt180703、wblxbt180704、wblxbt180705、wblxbt180706、wblxbt180707、wblxbt180708、wblxbt180709、wblxbt180710、wblxbt180711、wblxbt180712、wblxbt180713、wblxbt180714、wblxbt180715、wblxbt180716、wblxbt180717、wblxbt180718、wblxbt180719、wblxbt180720、wblxbt180721、wblxbt180722);
实施例1本发明检测方法用于王不留行标准汤剂高效液相特征图谱的检测
1对照品参照物溶液制备:取王不留行黄酮苷对照品、刺桐碱对照品、肥皂草苷对照品适量,精密称定,分别加70%甲醇制成每1ml含王不留行黄酮苷0.1mg、刺桐碱60μg、肥皂草苷50μg的溶液。再取原儿茶酸对照品、王不留行环肽a对照品、王不留行环肽b对照品、异牡荆素-2”-o-阿拉伯糖苷对照品适量,分别加甲醇制成每1ml含原儿茶酸60μg、王不留行环肽a60μg、王不留行环肽b60μg、异牡荆素-2”-o-阿拉伯糖苷30μg的溶液,即得。
2对照药材参照物溶液制备:取王不留行对照药材2.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
3供试品溶液制备:取王不留行标准汤剂约0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,超声处理(功率600w,频率40khz)20分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4检测:分别精密吸取对照品参照物溶液、对照药材参照物溶液、供试品溶液各10μl,注入液相色谱仪,测定,即得。
以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒径为5μm);乙腈为流动相a,以0.1%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;流速为每分钟1.0ml;柱温为30℃;检测波长为270nm。理论板数按王不留行黄酮苷峰计算应不低于3000。
5结果:供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)。
实施例2本发明检测方法用于王不留行配方颗粒高效液相特征图谱的检测
1对照品参照物溶液制备:配制方法同实施例1。
2对照药材参照物溶液制备:配制方法同实施例1。
3供试品溶液制备:取王不留行配方颗粒适量,研细,取1.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4检测:分别吸取对照品参照物溶液、对照药材参照物溶液和供试品溶液各10μl,注入高效液相色谱仪,色谱条件如下:
以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒径为5μm);乙腈为流动相a,以0.1%磷酸溶液为流动相b,梯度洗脱程序同实施例1;流速为每分钟1.0ml;柱温为30℃;检测波长为270nm。理论板数按王不留行黄酮苷峰计算应不低于3000。
5结果:供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)。
实施例3本发明检测方法用于王不留行药材高效液相特征图谱的检测
1对照品参照物溶液制备:配制方法同实施例1。
2对照药材参照物溶液制备:配制方法同实施例1。
3供试品溶液制备:取王不留行药材约2.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4检测:分别吸取对照品参照物溶液、对照药材参照物溶液和供试品溶液各10μl,注入高效液相色谱仪,色谱条件如下:
以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒度为5μm);乙腈为流动相a,以0.1%磷酸溶液为流动相b,梯度洗脱程序同实施例1;流速为每分钟1.0ml;柱温为30℃;检测波长为270nm。理论板数按王不留行黄酮苷峰计算应不低于3000。
5结果:供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)。
实施例4本发明检测方法用于王不留行饮片高效液相特征图谱的检测
1对照品参照物溶液制备:配制方法同实施例1。
2对照药参照物材溶液制备:配制方法同实施例1。
3供试品溶液制备:取王不留行饮片2.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4检测:分别吸取对照品参照物溶液、对照药材参照物溶液和供试品溶液各10μl,注入高效液相色谱仪,色谱条件如下:
以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒度为5μm);乙腈为流动相a,以0.1%磷酸溶液为流动相b,梯度洗脱程序同实施例1;流速为每分钟1.0ml;柱温为30℃;检测波长为270nm。理论板数按王不留行黄酮苷峰计算应不低于3000。
5结果:供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)。
实验例1色谱条件与系统适用性试验
流动相选择:考察了3种不同流动相的分离效果,分别为:(1)乙腈-0.1%甲酸梯度洗脱;(2)乙腈-0.1%甲酸梯度洗脱;(3)乙腈-0.1%磷酸梯度洗脱。结果表明,乙腈-0.1%磷酸梯度洗脱条件下色谱峰分离度、理论板数、对称性均更好,故将乙腈-0.1%磷酸梯度洗脱作为王不留行特征图谱测定方法的流动相。结果表明,在检测波长为270nm波长下各色谱峰峰形和对称性更好,整个色谱图信息量较大,故检测波长确定为270nm。与王不留行标准汤剂特征图谱方法一致。
波长选择:在以上拟定的实验条件基础上,利用二极管阵列检测器分别对王不留行黄酮苷对照品溶液、王不留行环肽a对照品溶液、王不留行环肽b对照品溶液、刺桐碱对照品溶液、肥皂草苷对照品溶液、异牡荆素-2”-o-阿拉伯糖苷对照品溶液、原儿茶酸对照品溶液、供试品溶液进行全波段扫描,并提取供试品溶液3d图以及在250nm、260nm、270nm、280nm波长下的色谱图。
柱温考察:在以上拟定的实验条件基础上,分别对柱温为25℃、30℃、35℃时进行考察。结果表明,在三种柱温条件下,各色谱峰都能较好分离,故参考王不留行标准汤剂特征图谱方法,暂定柱温为30℃进行后续考察。
流速考察:在以上拟定的实验条件基础上,分别对流速为0.8ml/min、1ml/min、1.2ml/min时进行了考察。结果表明,在三种流速条件下,各色谱峰都能较好分离,故参考王不留行标准汤剂特征图谱方法,暂定流速为1.0ml/min进行后续考察。
延迟性实验
在以上拟定的实验条件基础上,将色谱图采集时间延长至120min。
结果表明,在色谱图采集到60分钟后,仅在74分钟左右出现一个色谱峰,经过同等条件下进空针证实,此峰并非供试品原因产生。故当色谱图采集至60分钟时,已将色谱峰采集完全。将色谱图采集时间确定为60分钟。
综上所述,王不留行配方颗粒特征图谱色谱条件与系统适应性试验确定为:以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒径为5μm);以乙腈为流动相a,以0.1%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;流速为每分钟1.0ml;柱温为30℃;检测波长为270nm。理论板数按王不留行黄酮苷峰计算应不低于3000。见表9。
表9拟定的流动相梯度
实验例2供试品溶液的制备考察
提取方法考察:取本品(批号1804009)约1.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,分别对供试品提取方法为回流、超声时进行考察,提取时间30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
结果表明,对供试品分别进行超声提取与回流提取时效果一致。因超声提取操作更为简便,故供试品提取方法确定为超声提取。
提取溶剂考察:取本品(批号1804009)约1.0g,精密称定,置具塞锥形瓶中,分别对供试品提取溶剂为甲醇、70%甲醇、水时进行考察,溶剂加入量为50ml,密塞,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得。结果表明,水和70%甲醇提取效果较好,且无明显差异,暂定提取溶剂为70%甲醇。
提取时间考察:取本品(批号1804009)约1.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,超声处理(功率600w,频率40khz),分别对供试品提取时间为20分钟、30分钟、40分钟时进行考察,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
结果表明,在三种提取时间处理供试品时,无明显差异,暂定供试品提取时间确定为30分钟。
综上所述,王不留行配方颗粒特征图谱供试品溶液的制备方法确定为:取本品适量,研细,取约1.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
实验例3精密度试验
取王不留行配方颗粒(批号:1804009)约1.0g,精密称定1份,按拟定实验方法进行制备及测定,连续进样6次,每次10μl,进行测定。结果表明,各特征峰保留时间的rsd在0.03%-0.13%,各特征峰相对峰面积的rsd在0.07%-0.84%,该仪器精密度良好。
实验例4重复性试验
精密称取王不留行标准汤剂冻干粉(批号:dqybt180401)6份,按拟定实验方法进行制备及测定。结果表明,各特征峰相对保留时间的rsd为0.02%~1.11%,各特征峰相对峰面积的rsd在4.90%~10.66%。该方法重复性良好。结果表明,各特征峰相对保留时间的rsd在0.00%-0.05%,各特征峰相对峰面积的rsd在0.00%-5.00%。
实验例5色谱柱耐用性考察
在以上拟定的实验条件基础上,分别对色谱柱为zorbaxeclipsexdb-c18analytical4.6×250mm5-micron(安捷伦色谱柱)、diamonsil5μmc18(2)250×4.6mm(迪马色谱柱)、shim-packgist5μmc184.6×250mm(岛津色谱柱)时进行考察。结果表明,用上述3种色谱柱对样品进行检测,各特征峰相对保留时间的rsd在0.20%~7.65%;各特征峰相对峰面积的rsd在0.00%~12.01%。
实验例6稳定性试验
在以上拟定的实验条件基础上,取同一供试品溶液,分别于0h,2h,4h,8h,16h,24h测定。结果表明,各特征峰保留时间的rsd在0.01%~0.12%,各特征峰峰面积的rsd在0.10%~0.46%。该方法供试品在24小时内稳定性良好。
实验例7王不留行样品对照图谱的建立
1.王不留行标准汤剂特征图谱的建立
采用最终确定的分析方法,对本品22批王不留行标准汤剂样品(四川新绿色药业科技发展有限公司制备,批号:wblxbt180701、wblxbt180702、wblxbt180703、wblxbt180704、wblxbt180705、wblxbt180706、wblxbt180707、wblxbt180708、wblxbt180709、wblxbt180710、wblxbt180711、wblxbt180712、wblxbt180713、wblxbt180714、wblxbt180715、wblxbt180716、wblxbt180717、wblxbt180718、wblxbt180719、wblxbt180720、wblxbt180721、wblxbt180722)进行特征图谱的测定,计算相对保留时间、相对峰面积。
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了11个重复性较好的峰作为特征峰。结果表明,当峰4作为s峰时,22批次王不留行标准汤剂特征峰相对保留时间rsd均小于2.0%。最终规定:王不留行标准汤剂供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)。
采用中药色谱指纹图谱相似度评价系统(2012版)对22批王不留行标准汤剂特征图谱进行合成,建立了王不留行标准汤剂特征图谱的对照图谱,建立的特征图谱检测方法可以较为准确地整体控制王不留行标注汤剂的质量。
2.王不留行配方颗粒特征图谱的建立
采用拟定的方法对王不留行配方颗粒3批样品(批号:sy1807001、sy1807002、sy1807003)进行特征图谱的测定,计算相对保留时间、相对峰面积和相对峰高。
结果表明,3批次王不留行配方颗粒11个特征峰相对保留时间rsd均小于2.0%。最终规定:供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)。
采用中药色谱指纹图谱相似度评价系统(2012版)对3批次王不留行配方颗粒特征图谱进行合成,建立了王不留行配方颗粒特征图谱的对照图谱,建立的特征图谱检测方法可以较为准确地整体控制王不留行配方颗粒的质量。
3.王不留行药材特征图谱的建立
采用最终确定的分析方法,对本品22批王不留行药材(四川新绿色药业科技发展有限公司,批号:xls201805338、xls201805339、xls201805340、xls201805341、xls201805343、xls201805344、010458-1706001、xls201806513、xls201806514、xls201806515、xls201806516、xls201806517、xls201806563、xls201806564、xls201806565、xls201806566、xls201806567、xls201806568、xls201806569、xls20180670、xls201806571、xls201806572)进行特征图谱的测定,计算相对保留时间、相对峰面积。
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了11个重复性较好的峰作为特征峰。结果表明,当峰4作为s峰时,22批次王不留行药材特征峰相对保留时间rsd均小于2.0%。最终规定:王不留行药材供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)
采用中药色谱指纹图谱相似度评价系统(2012版)对22批王不留行药材特征图谱进行合成,建立了王不留行药材特征图谱的对照图谱,建立的特征图谱检测方法可以较为准确地整体控制王不留行药材的质量。
4.王不留行饮片特征图谱的建立
采用最终确定的分析方法,对本品22批王不留行饮片(四川新绿色药业科技发展有限公司,批号:wblx180701、wblx180702、wblx180703、wblx180704、wblx180705、wblx180706、wblx180707、wblx180708、wblx180709、wblx180710、wblx180711、wblx180712、wblx180713、wblx180714、wblx180715、wblx180716、wblx180717、wblx180718、wblx180719、wblx180720、wblx180721、wblx180722)进行特征图谱的测定,计算相对保留时间、相对峰面积。
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了11个重复性较好的峰作为特征峰。结果表明,当峰4作为s峰时,22批次王不留行标准汤剂特征峰相对保留时间rsd均小于2.0%。最终规定:王不留行饮片供试品特征图谱中应呈现11个特征峰,并应与对照药材参照物色谱中的11个特征峰保留时间相对应,其中7个峰应分别与相应的参照物峰保留时间相同,与王不留行黄酮苷参照物峰相应的峰为s峰,除峰2、峰3、峰10、峰11以外,计算其余各特征峰与s峰的相对保留时间,其中峰1的相对保留时间在规定值的±10%之内,峰4~9的相对保留时间应在规定值的±5%之内。规定值为:0.208(峰1)、1.000(峰4s)、1.050(峰5)、1.126(峰6)、1.210(峰7)、1.439(峰8)、1.513(峰9)
采用中药色谱指纹图谱相似度评价系统(2012版)对22批王不留行饮片特征图谱进行合成,建立了王不留行饮片特征图谱的对照图谱,建立的特征图谱检测方法可以较为准确地整体控制王不留行饮片的质量。
实验例8本发明检测方法用于王不留行药材、饮片、标准汤剂高效液相特征图谱的对比
1王不留行药材供试品溶液的制备:取22批次王不留行药材各2.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,即得。
2王不留行饮片供试品溶液的制备:取22批次王不留行饮片各2.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,称定重量,超声处理(功率600w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,即得。
3王不留行标准汤剂供试品溶液的制备:取22批次王不留行标准汤剂各0.5g,实施例1相同方法配制。
4王不留行配方颗粒供试品溶液的制备:取3批次王不留行配方颗粒各1g,实施例2相同方法配制。
5检测:分别吸取王不留行药材、饮片、标准汤剂、配方颗粒供试品溶液各10μl,注入高效液相色谱仪,色谱条件如下:
以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒径为5μm);乙腈为流动相a,以0.1%磷酸溶液为流动相b,梯度洗脱程序同实施例1;流速为每分钟1.0ml;柱温为30℃;检测波长为270nm。理论板数按王不留行黄酮苷峰计算应不低于3000。
6结果:采用中药色谱指纹图谱相似度评价系统(2012版)分别将22批王不留行药材特征图谱合成一张王不留行药材对照图谱,22批王不留行饮片特征图谱合成一张饮片对照图谱,22批王不留行标准汤剂特征图谱合成一张标准汤剂对照图谱,3批王不留行配方颗粒特征图谱合成一张配方颗粒对照图谱。将以上王不留行药材、饮片、标准汤剂、配方颗粒的对照图谱进行对比。结果表明,王不留行药材、饮片、标准汤剂、配方颗粒中均能检出11个特征峰,其物质基础一致,本发明方法可以准确高效地检测出王不留行药材、饮片、标准汤剂、配方颗粒中的特征成分,实现整体控制王不留行药材、饮片、标准汤剂、配方颗粒质量的目的。
综上,本发明的王不留行药材、饮片、标准汤剂、配方颗粒高效液相特征图谱的检测方法,能整体控制王不留行药材、饮片、标准汤剂、配方颗粒中的特征成分,确保王不留行药材、饮片、标准汤剂、配方颗粒质量的整体稳定,且方法操作简单,精密度高,稳定性好,重复性好,准确度高。
- 还没有人留言评论。精彩留言会获得点赞!