一种烟用香精香料的气相色谱-串联质谱分析方法与流程
本发明属于烟用香精香料成分检测技术领域,具体涉及一种超纯水稀释、有机溶剂提取、多壁碳纳米管净化、气相色谱-串联质谱(gc-ms/ms)同时测定烟用香精香料中345种香味成分的分析方法。
背景技术:
烟用香精香料是卷烟生产中必不可少的物质,既可以赋予卷烟烟气特定的吸味,弥补和增强卷烟烟气的香气,又可使卷烟烟气香味柔和细腻,其配方是卷烟行业的核心技术,对改善卷烟的口感,突出卷烟风格具有重要的作用。因此得到系统而全面的烟用香精香料感官关键成分分析数据,是进行卷烟设计的基础和依据,获得正确的烟用香精香料关键成分的定性、定量分析数据对仿香、创香至关重要。
目前烟用香精香料香味成分的检测主要基于非靶标gc/ms方法,首先进行全扫描(scan)分析,通过谱库检索定性,因此仅能测定可准确定性的化合物。受样品基质干扰、gc/ms灵敏度不足等分析方法问题制约,既往研究覆盖的香味成分数量较为有限,仅能覆盖几十种。而且,目前的方法并非用标准曲线定量,而是采用半定量的方法,即用各化合物的峰面积与内标的峰面积比例计算。因此,迫切需要建立一种烟用香精香料中关键香味成分超多靶标、操作简单、准确快速、高通量、高灵敏、高效率、绝对定量的分离分析方法。
技术实现要素:
本文发明的目的旨在寻找操作简单、通用性强、成本低、环境友好、能满足性质差异较大的多种目标物的同时提取的通用型和高效型的烟用香精香料样品前处理方法,并结合气相色谱-串联质谱技术,寻找最优色谱条件及mrm(多反应监测模式)参数,实现对烟用香精香料中超多靶标香味成分的检测。该方法快速、准确、灵敏、成本低、易操作、通量高,可同时对345种香味成分进行测定,可满足对大批量样品进行快速分析检测的需要。
本发明的目的是通过以下技术方案来实现的:一种检测烟用香精香料中多种香味成分的方法,首先向待测烟用香精香料样品中加入超纯水稀释,有机溶剂提取,然后在盐析作用下,用干燥剂除水,再以多壁碳纳米管作为逆固相分散吸附剂,对样品进行净化,结合气相色谱-串联质谱联用技术实现对烟用香精香料中多种香味成分的同时检测,具体步骤如下:
1)样品提取:先将样品加超纯水涡旋,再加入提取溶剂及内标工作液,涡旋,之后加入氯化钠,涡旋、离心;
具体过程为:称取2g烟用香精香料样品于50ml具塞离心管中,加入2~5ml超纯水,涡旋1~5min;加入10ml提取溶剂及内标工作液,以2500r/min涡旋1~2min,而后加入1.5~3.5g氯化钠,以2500r/min涡旋1~2min,5000~8000r/min离心3~5min;
2)样品净化:取步骤1)最后离心的上清液,加入无水硫酸镁和多壁碳纳米管,涡旋、离心,上清液过滤后待测;
具体过程为:取1~1.5ml上清液于2ml离心管中,加入150~200mg无水硫酸镁和5~10mg多壁碳纳米管,立即以2500r/min涡旋1~2min,5000~8000r/min离心3~5min;上清液经0.22μm有机相滤膜过滤后待测;
(3)样品检测:将待测液用气相色谱-串联质谱分析,采用基质匹配标准工作溶液制作标准曲线,用标准曲线定量;
gc-ms/ms分析条件如下:
色谱柱:弹性石英毛细管色谱柱,固定相为5%苯基-甲基聚硅氧烷,规格60m×0.25mm×0.25μm,进样口端串联预柱(5m×0.25mm);进样口温度:280℃;进样量:0.8~1.0μl;进样方式:不分流进样,不分流时间1min;载气:氦气,恒流模式,流速1.5ml/min;程序升温;电离模式:电子轰击电离,电离能70ev;灯丝电流:35μa;离子源温度:280℃;四级杆温度:150℃;传输线温度:280℃;q2碰撞气:氮气(纯度99.999%),流量1.5ml/min;淬灭气:氦气(纯度为99.999%),流量2.25ml/min;扫描方式:多反应监测(mrm)模式。
本发明中,所述的345种香味成分是通过对美欧烟草添加剂名单、bat烟草添加剂名单、中国烟草制品许可使用添加剂名单的整合、梳理和筛选得到的。
本发明中,所述的香味成分包括醛、酮、醇、酚、醚、酯、内酯、烯、吡啶、吡咯、吡嗪、硫化物、酰胺、酰亚胺等多类成分。
本发明中,所述的提取溶剂为乙腈,提取溶剂还可使用二氯甲烷。
本发明中,所述的内标为d8-苯乙酮,配成浓度为30mg/l的d8-苯乙酮乙腈溶液,每个样品的加入量为80μl;内标还可使用苯己酮、苯戊酮、4-溴苯戊酮、丙酸-2-苯乙酯、3-丙酸苯乙酯、氘代萘、蒽、苯并芘。
本发明中,所述的多壁碳纳米管为:外径10~20nm,长度10~20μm,比表面积>165m2/g,纯度>95%。
本发明中,所述的基质匹配标准工作溶液的配制方法如下:将香精香料样品按照相同的前处理方式处理后作为基质提取液,但提取时不添加内标,用该基质提取液稀释溶剂标准工作溶液,加入的待稀释溶剂标准工作溶液的体积不超过总体积的5%。
本发明中,所述的标准曲线定量是根据样品的性质,选择标准加入法、内标法等方法建立标准工作曲线,然后根据检测结果及各目标物的标准曲线计算相应成分的含量。
本发明中,所述的预柱的作用如下:减少分析柱前端的污染,延长柱寿命;有助于在柱前端聚焦样品,以获得更好的峰形。
本发明中,所述的gc-ms/ms分析条件中的程序升温过程如下:初始温度75℃,保持5min后以1℃/min升至150℃,保持1min,然后以2℃/min升至260℃,保持1min,最后以10℃/min升至280℃,保持10min。
本发明中,所述的gc-ms/ms分析条件中的mrm参数包括保留时间的确定,母离子、子离子和碰撞能量的选择优化。首先将各化合物进行全扫描(fullscan)分析(扫描范围m/z20~330),确定保留时间及一级质谱图,并筛选2~4个质荷比及丰度较大的离子作为备选母离子;再将上述各母离子在不同的碰撞能量下(5、10、15、20、25、30、35、40ev)进行产物离子扫描(productionscan),每个化合物筛选出4~8对离子对及最优碰撞能量;最后,用mrm模式对标准溶液、基质提取液、添加标准品的基质提取液进行分析,选择抗干扰能力强、灵敏度高的两对离子对分别作为定量及定性离子对。目标物的mrm参数如表1所示。
表1目标物及其内标的mrm参数
与现有技术相比,本发明方法具有如下优良效果:
(1)目前烟用香精香料成分的分析集中在气相色谱质谱法(gc/ms)上,即,前处理完成后,首先进行全扫描(scan)分析,通过检索标准质谱图库定性,受样品基质干扰、gc/ms灵敏度不足等分析方法问题制约,能准确定性的化合物数量极为有限,仅为几十种。本发明用气相色谱-串联质谱(gc-ms/ms)可同时测定烟用香精香料中345种香味成分,和以往方法相比可分析化合物范围更广、数量更多;每个化合物都选择优化了相应的定量离子对和定性离子对,无需标准谱库检索,化合物的定性更准确;且方法灵敏度更高、精密度和重复性更好。
(2)目前的气相色谱质谱法是将全扫描分析中能准确定性的化合物用选择离子监测(sim)定量,定量方式为用各化合物选择离子的峰面积与内标选择离子峰面积比例作为该化合物响应值,是半定量方法。而本发明用基质匹配标准工作液建立标准曲线进行绝对定量,因此本发明的准确度更高。
(3)基质效应是气相色谱质谱分析时普遍存在的问题,主要表现为基质增强效应,即基质成分的存在减少了色谱系统活性位点与待测物分子作用的机会,从而造成待测物信号有所增强。基质溶液的ph值、共提物的种类和数量均会对待测物的响应产生影响。本发明采用基质匹配标准工作液可以校正基质效应引入的定量误差,定量结果更准确。
(4)本发明用乙腈或二氯甲烷提取、氯化钠盐析、无水硫酸镁除水、多壁碳纳米管分散固相萃取净化,操作简单快速,通量高,成本低,溶剂用量少,环境友好,能满足性质差异较大的多种目标物的同时提取、净化。
(5)本发明用多壁碳纳米管作为逆固相分散吸附剂,多壁碳纳米管是由多层碳石墨片卷曲而成的无缝中空管,具有特殊的物理化学性质、独特的结构和巨大的比表面积,对杂质具有很强的吸附能力,净化效果好,在降低基质效应的同时,亦排除了常用的吸附剂如psa、gcb、c18对目标分析物吸附而造成的影响,并提供更干净的上机溶液,减少了仪器在使用过程中的维护。
附图说明
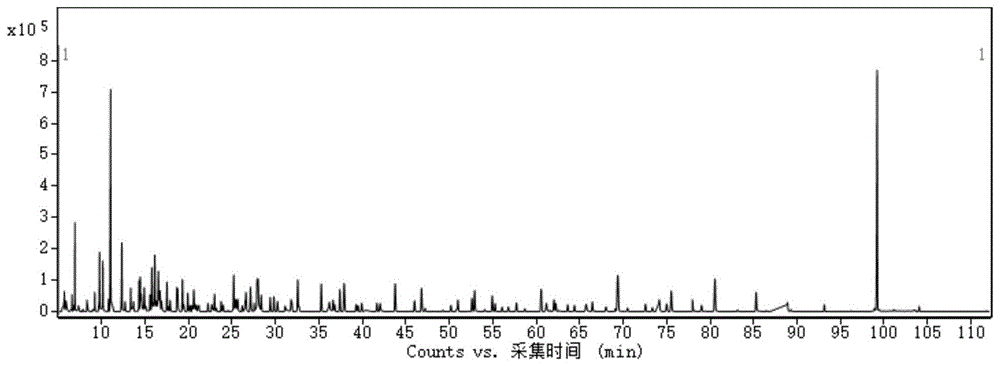
图1标准溶液在gc-ms/ms上的总离子流图。
具体实施方式
本发明以下结合实例做进一步描述,但并不是限制本发明。
实例1:
称取2g烟用香精香料样品于50ml具塞离心管中,加入4ml水,涡旋2min;加入10ml乙腈以及80μl30.0mg/ld8-苯乙酮内标工作液,以2500r/min涡旋2min,而后加入2.5g氯化钠,再以2500r/min涡旋1min,8000r/min离心3min;取1ml上清液于2ml离心管中,加入150mg无水硫酸镁和5mg多壁碳纳米管,立即以2500r/min涡旋2min,8000r/min离心3min;上清液经0.22μm有机相滤膜过滤后进行gc-ms/ms分析;
gc-ms/ms分析条件如下:
气相色谱条件:色谱柱:弹性石英毛细管色谱柱,固定相为50%苯基-甲基聚硅氧烷,规格60m×0.25mm×0.25μm,进样口端串联预柱(5m×0.25mm);进样口温度:280℃;进样量:0.8μl;进样方式:不分流进样,不分流时间1min;载气:氦气,恒流模式,流速1.5ml/min;程序升温:初始温度75℃,保持5min后以1℃/min升至150℃,保持1min,然后以2℃/min升至260℃,保持1min,最后以10℃/min升至280℃,保持10min。
质谱条件:电离模式:电子轰击电离,电离能70ev;灯丝电流:35μa;离子源温度:280℃;四级杆温度:150℃;传输线温度:280℃;q2碰撞气:氮气(纯度99.999%),流量1.5ml/min;淬灭气:氦气(纯度为99.999%),流量2.25ml/min;扫描方式:多反应监测(mrm)模式。mrm参数见表1。
标准工作溶液的浓度分别为0.01,0.02,0.05,0.1,0.2,0.5和1μg/ml。分别对这些标准溶液进行gc-ms/ms检测,以目标物峰面积与内标峰面积的比对目标物浓度进行线性回归分析,各标准曲线的线性关系良好。进行了0.1和1μg/g两个水平的添加回收试验,目标物在两个添加水平下平均回收率在66.7~126.7%之间,rsd在0.5~17.4%之间。以3倍信噪比和10倍信噪比计算方法检出限(lod)和定量限(loq),所有目标物的检出限在0.4~35ng/g之间,定量限在1~117ng/g之间,其中有312个化合物的定量限在1~50ng/g之间,33个化合物的定量限在51~117ng/g之间。方法表征数据见表2。结果表明,该方法具有良好的回收率,精密度、灵敏度、稳定性较好,可满足分析检测需要。
表2345种目标物的相关系数、回收率(n=5)、相对标准偏差、检测限和定量限
实例2:
称取2g烟用香精香料样品于50ml具塞离心管中,加入标准品溶液,使目标物添加水平为0.5μg/g,加入4ml水,涡旋2min;加入10ml乙腈以及80μl30.0mg/ld8-苯乙酮内标工作液,以2500r/min涡旋2min,而后加入2.5g氯化钠,再以2500r/min涡旋1min,8000r/min离心3min;取1ml上清液于2ml离心管中,加入150mg无水硫酸镁和5mg多壁碳纳米管,立即以2500r/min涡旋2min,8000r/min离心3min;上清液经0.22μm有机相滤膜过滤后进行gc-ms/ms分析;gc-ms/ms分析条件参考实例1。
按照上述操作进行日内5次平行和日间5次平行试验,测定结果分别为日内精密度和日间精密度,如表3所示,日内精密度和日间的精密度分别为0.5~10.7%、0.7~10.0%。结果表明,该方法具有良好的精密度,稳定性较好,可满足分析检测需要。
表3345种目标物的日内精密度和日间精密度
实例3:
用实例1的方法检测了5个不同的烟用香精香料样品,共检出254种目标物。其中,1号样品共检出162种目标物,含量最高的是3-甲基-2,5-呋喃二酮,其次依次是糠醛、3-糠醛、苯甲醛和乳酸乙酯;2号样品共检出189种目标物,含量最高的是甲基环戊烯醇酮,其次依次是丁香酚、3-甲基-2,5-呋喃二酮、乳酸乙酯和茄酮;3号样品共检出176种目标物,含量最高的是乳酸乙酯,其次依次是3-甲基-2,5-呋喃二酮、茄酮、壬酸乙酯和α-当归内酯;4号样品共检出254种目标物,含量最高的是茄酮,其次依次是3-甲基-2,5-呋喃二酮、巨豆三烯酮、棕榈酸乙酯和油酸乙酯;5号样品共检出253种目标物,含量最高的是茄酮,其次依次是巨豆三烯酮、3-甲基-2,5-呋喃二酮、对乙烯基愈创木酚和2(5h)-呋喃酮。
- 还没有人留言评论。精彩留言会获得点赞!