一种苏黄止咳胶囊指纹图谱的检测方法及其应用与流程
本发明涉及但不限于药物分析技术,尤指但不限于一种苏黄止咳胶囊指纹图谱的检测方法及其应用。
背景技术:
苏黄止咳胶囊的处方由密炙麻黄、紫苏叶、地龙、炙枇杷叶、紫苏子、蝉蜕、前胡、牛蒡子、五味子组成。其中麻黄、地龙、蝉蜕、紫苏叶、前胡为其疏风、散风之药,用以止咳、利咽、止痒;紫苏子、五味子解痉缓急;牛蒡子、枇杷叶润肺清热。诸药合用,辛温宣肺,疏风解痉,通窍降气,豁痰平喘,使风散痰消挛解,肺气得以宣降,气机通畅,最终达到风咳止咳整体调整、标本兼治的目的,用于治疗咳嗽变异型哮喘和感冒后咳嗽。临床试验表明该药的效果良好,且不良反应也较目前常用的西药小。
中国专利申请cn104007222a公开了一种苏黄止咳胶囊的检测方法,用于苏黄止咳胶囊的质量控制;该方法采用薄层色谱法对苏黄止咳胶囊中麻黄、紫苏叶、地龙、牛蒡子、五味子五味药材进行定性鉴别;采用高效液相色谱法对苏黄止咳胶囊中盐酸麻黄碱的含量进行测定(用十八烷基硅烷键合硅胶为填充剂;以乙腈-水-磷酸-三乙胺1.5:98.3:0.1:0.1为流动相;检测波长为205nm)。
中国专利申请cn108490083a公开了一种苏黄止咳胶囊的质量检测方法,该检测方法包括麻黄碱和伪麻黄碱(以极性乙醚连接苯基键合硅胶为填充剂;以甲醇-0.092%磷酸溶液(含0.125%三乙胺)(1.5∶98.5)为流动相;检测波长为210nm)以及牛蒡苷(以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(1∶1.1)为流动相;检测波长为280nm)的含量测定方法,同时对苏黄止咳胶囊中盐酸麻黄碱和盐酸伪麻黄碱进行质量检测的方法。
尽管现有技术公开了盐酸麻黄碱、盐酸伪麻黄碱以及牛蒡苷的质量检测方法,但是没有苏黄止咳胶囊指纹图谱的检测方法,同时,也尚未确证建立有效成分间的关系。
技术实现要素:
以下是对本文详细描述的主题的概述。本概述并非是为了限制权利要求的保护范围。
本申请提供了一种苏黄止咳胶囊指纹图谱的检测方法及其应用,不仅确立了苏黄止咳胶囊质量控制的标记物,同时可以更好地控制产品的质量。
本申请提供了一种苏黄止咳胶囊指纹图谱的检测方法,所述的检测方法包括如下步骤:
(1)供试品溶液的制备:
取苏黄止咳胶囊,去胶囊壳,精密称定0.5g,置具塞锥形瓶中,精密加入30±5ml60±10体积%甲醇水溶液称定重量,超声40±10min,放冷至室温,补足减失重量,滤过,取续滤液,过0.45μm微孔滤膜,即得供试品溶液;
(2)hplc色谱条件的确立
色谱柱:采用填料为十八烷基硅烷键合硅胶的色谱柱,可选规格4.6×250mm,5μm;
流动相:流动相a和流动相b的混合物,其中,流动相a为0.1±0.05体积%甲酸水溶液,流动相b为乙腈;流动相梯度洗脱;
柱温:30±5℃,优选地,28℃;
流速:1±0.5ml·min-1,优选地,1ml·min-1;
检测波长:230±30nm,优选地,254nm;
进样量:10±5μl,优选地,10μl;
(3)hplc图谱的测定
取所述供试品溶液,根据所述hplc色谱条件进样,得到hplc色谱图;
(4)标准指纹图谱的建立
在上述hplc图谱中确定共有特征峰,优选地,确立22个共有特征峰,所述共有特征峰包括下列成分的共有特征峰:
腺苷、1-咖啡酰奎宁酸、绿原酸、夏佛塔苷、牛蒡苷、五味子醇甲、戈米辛d、五味子醇乙、当归酰戈米辛h、白花前胡甲素和五味子乙素。
在本申请的实施方案中,腺苷、1-咖啡酰奎宁酸、绿原酸、夏佛塔苷、牛蒡苷、五味子醇甲、戈米辛d、五味子醇乙、当归酰戈米辛h、白花前胡甲素和五味子乙素为11个共有峰,并经对照品指认,其相对保留时间如下:分别以19.6417min、51.5968min和103.2624min的色谱峰保留时间为参照,相对保留时间分别为:以19.6417min色谱峰保留时间为参照,0.3207、0.7817;以51.5968min的色谱峰保留时间为参照,0.6455、0.9116、1.3868;以103.2624min的色谱峰保留时间为参照,1.0000、1.0374、1.0663、1.1281、1.1975、1.3501。
在本申请的实施方案中,所述22个共有特征峰的相对保留时间为:选择三个不同时间(19.6417min、51.5968min和103.2624min)的色谱峰保留时间为参照计算其余19个共有峰相对保留时间,其中22个共有峰保留时间分别为:0.3207、0.3605、0.4805、0.5080、0.7817、1.0000(19.6417min作为参考);
0.5141、0.6455、0.7566、0.9116、1.0000、1.3868、1.4096(51.5968min作为参考);
0.9580、1.0000、1.0374、1.0663、1.1281、1.1975、1.3234、1.3501、1.3561(103.2624min作为参考)。
在本申请的实施方案中,所述流动相洗脱梯度为:
0~15min,0.1±0.05体积%甲酸水:乙腈为96:4;
15~40min,0.1±0.05体积%甲酸水:乙腈为96~85:4~15;
40~90min,0.1±0.05体积%甲酸水:乙腈为85~65:15~35;
90~110min,0.1±0.05体积%甲酸水:乙腈为65~50:35~50;
110~130min,0.1±0.05体积%甲酸水:乙腈为50~40:50~60;
130~140min,0.1±0.05体积%甲酸水:乙腈为40~10:60~90;
140~145min,0.1±0.05体积%甲酸水:乙腈为10~5:90~95。
在本申请的优选实施方案中,本申请提供的一种苏黄止咳胶囊指纹图谱的检测方法,其中,hplc色谱条件为:
色谱柱:采用填料为十八烷基硅烷键合硅胶的色谱柱,可选规格4.6×250mm,5μm;
流动相:流动相a和流动相b的混合物,其中,流动相a为0.05体积%甲酸水溶液,流动相b为乙腈;流动相梯度洗脱;
柱温:28℃;
流速:1ml·min-1;
检测波长:254nm;
进样量:10μl。
在本申请的优选实施方案中,所述供试品溶液的制备为:
取苏黄止咳胶囊,去胶囊壳,精密称定0.5g,置具塞锥形瓶中,精密加入30ml60体积%甲醇水溶液称定重量,超声40min,补足减失重量,滤过,取续滤液,过0.45μm微孔滤膜,即得供试品溶液;
所述流动相洗脱梯度为:
0~15min,0.05体积%甲酸水:乙腈为96:4;
15~40min,0.05体积%甲酸水:乙腈为96~85:4~15;
40~90min,0.05体积%甲酸水:乙腈为85~65:15~35;
90~110min,0.05体积%甲酸水:乙腈为65~50:35~50;
110~130min,0.05体积%甲酸水:乙腈为50~40:50~60;
130~140min,0.05体积%甲酸水:乙腈为40~10:60~90;
140~145min,0.05体积%甲酸水:乙腈为10~5:90~95。
在本申请的实施方案中,在确定了标准指纹图谱后,还包括采用中国国家药典委员会的“中药色谱指纹图谱相似度评价系统软件(2012版)”进行评价。
另一方面,本申请提供了上述检测方法得到的指纹图谱在苏黄止咳胶囊质量控制中的应用。
所建立指纹图谱可系统监测苏黄止咳胶囊整体质量,同时所建立指纹图谱指认共有成分中白花前胡甲素、五味子醇甲及伪麻黄碱,牛蒡苷代谢产物牛蒡苷元具明显的抗炎活性。可在控制生产稳定苏黄止咳胶囊同时保证苏黄药效的稳定性。
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在说明书、权利要求书以及附图中所特别指出的结构来实现和获得。
附图说明
附图用来提供对本发明技术方案的进一步理解,并且构成说明书的一部分,与本申请的实施例一起用于解释本发明的技术方案,并不构成对本发明技术方案的限制。
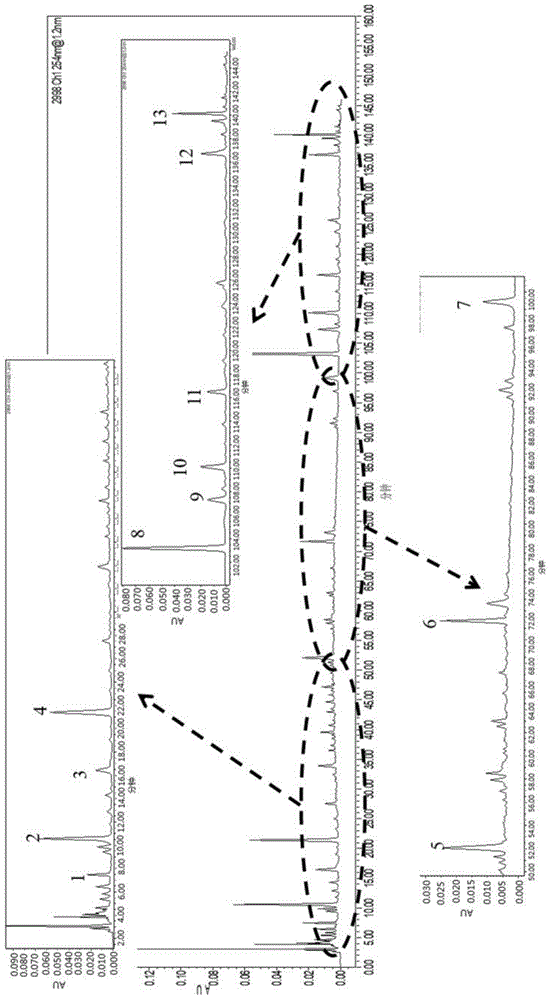
图1为实施例1制备得到的苏黄止咳胶囊指纹图谱色谱条件优化后谱图;
图2为实施例1制备得到的苏黄止咳胶囊精密度试验图谱;
图3为实施例1制备得到的苏黄止咳胶囊稳定性试验图谱;
图4为实施例1制备得到的苏黄止咳胶囊重复性试验图谱;
图5为实施例1制备得到的苏黄止咳胶囊对照指纹图谱;
图6为实施例1制备得到的16批次苏黄止咳胶囊指纹图谱;
图7为对比例中不同流动相组成的苏黄止咳胶囊hplc图谱;
图8为对比例中不同流动相洗脱梯度的苏黄止咳胶囊hplc图谱;
图9为对比例中不同酸的苏黄止咳胶囊hplc图谱;
图10为对比例中不同色谱柱的苏黄止咳胶囊hplc图谱;
图11为对比例中不同波长的苏黄止咳胶囊hplc图谱;
图12为对比例中不同流速的苏黄止咳胶囊hplc图谱;
图13为对比例中不同柱温的苏黄止咳胶囊hplc图谱。
具体实施方式
为使本申请的目的、技术方案和优点更加清楚明白,下文中将对本发明的实施例进行详细说明。需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互任意组合。
仪器:waters1525(含waters2707auto-sampler、waters2489photodiodearraydetector、waters1525binaryhplcpump)和watersacquityarc高效液相色谱仪(含samplemanagerftn-r、waters2998photodiodearraydetector、quaternarysolventmanager-r)、色谱柱:welch(
材料试剂:乙腈(分析纯上海星可生化高纯溶剂有限公司);甲醇(分析纯上海星可生化高纯溶剂有限公司);甲酸(分析纯上海申博化工有限公司)。苏黄止咳胶囊(扬子江药业集团北京海燕药业有限公司,批号:15121111)
实施例1
样品溶液的制备:取苏黄止咳胶囊,去胶囊壳,精密称定0.5g,置具塞锥形瓶中,精密加入30ml60体积%甲醇水溶液称定重量,超声40min,放冷至室温,补足减失重量,滤过,取续滤液,过0.45μm微孔滤膜,既得供试品溶液,备用。
1.色谱条件对比
该部分对影响色谱行为的影响因素如流动相组成、流动相洗脱梯度、不同酸、不同酸浓度、不同色谱柱、不同检测波长、不同流速及不同柱温进行考察优化最佳色谱条件。结果确定色谱条件为:采用welch(
2.供试品制备条件的对比
该部分以谱图中峰面积占总峰面积大于2%的13个色谱峰面积为评价指标,对影响苏黄止咳胶囊内容物提取的影响因素如提取方法、提取溶剂、提取时间、提取次数、料液比及甲醇浓度考察进行优化最佳提取条件。结果确定提取条件为:采用超声提取方式,提取时间为40min,一次,料液比(v/m)1:60,提取溶剂为60体积%甲醇。
3.方法学验证
指纹色谱图以单个峰面积大于1%,总峰面积大于40%的22个色谱峰为参照,用于评价建立方法的可行性。
3.1精密度试验
供试品溶液的制备:取苏黄止咳胶囊去除胶囊壳,充分混匀,精密称取0.5g苏黄止咳胶囊粉末,置于50ml具塞锥形瓶中,精密加入30ml60%甲醇溶液,称定重量,超声提取40min,放冷,用相应的60%甲醇溶液补足减失重量,滤过,取续滤液,滤液过0.45μm微孔滤膜,既得供试品溶液。连续6次进样,考察不同进样次数的谱图中主要色谱峰相对峰面积及其相对保留时间的一致性,结果得精密度图谱,见附图2。
以图谱中峰面积占总峰面积大于1%的22个色谱峰为参照,分别以保留时间为19.6417min,51.5968min及103.2624min三个色谱峰保留时间和峰面积为参照计算其余色谱峰相对保留时间和相对峰面积结果,见表1、2。表1指纹图谱精密度试验相对保留时间结果
表2指纹图谱精密度试验相对峰面积结果
结果各色谱峰相对保留时间和相对峰面积rsd值均小于3%,表明仪器精密度良好。
3.2稳定性试验
供试品溶液的制备:取苏黄止咳胶囊去除胶囊壳,充分混匀,精密称取0.5g苏黄止咳胶囊粉末,置于50ml具塞锥形瓶中,精密加入30ml60%甲醇溶液,称定重量,超声提取40min,放冷,用相应的60%甲醇溶液补足减失重量,滤过,取续滤液,滤液过0.45μm微孔滤膜,既得供试品溶液。用该样品分别在样品提取0、4、8、12、16、24、48h时进样,考察样品在不同时间段的谱图中主要色谱峰相对峰面积及其相对保留时间的一致性,结果稳定性图谱,见附图3。
以图谱中峰面积占总峰面积大于1%的22个色谱峰为参照,分别以保留时间为19.6417min,51.5968min及103.2624min三个色谱峰保留时间和峰面积为参照计算其余色谱峰相对保留时间和相对峰面积结果,见表3、4。
表3指纹图谱稳定性试验相对保留时间结果
表4指纹图谱稳定性试验相对峰面积结果
结果,各色谱峰相对保留时间和相对峰面积rsd值均小于3%,表明供试品溶液在48h内稳定性良好。
3.3重复性试验
供试品溶液的制备:取苏黄止咳胶囊去除胶囊壳,充分混匀,精密称取0.5g苏黄止咳胶囊粉末6份,分别置于50ml具塞锥形瓶中,精密加入30ml60%甲醇溶液,称定重量,超声提取40min,放冷,用相应的60%甲醇溶液补足减失重量,滤过,取续滤液,滤液过0.45μm微孔滤膜,既得供试品溶液。分别进样,考察重复样品的谱图中主要色谱峰相对峰面积及其相对保留时间的一致性,结果稳定性图谱,见附图4。
以图谱中峰面积占总峰面积大于1%的22个色谱峰为参照,分别以保留时间为19.6417min,51.5968min及103.2624min三个色谱峰保留时间和峰面积为参照计算其余色谱峰相对保留时间和相对峰面积结果,见表5、6。
表5指纹图谱重复性试验相对保留时间结果
表6指纹图谱重复性试验相对峰面积结果
结果,各色谱峰相对保留时间和相对峰面积rsd值均小于3%,表明方法重现性良好。
方法学验证结果表明,以图谱中峰面积占总峰面积大于1%的22个色谱峰为参照,分别以保留时间为19.6417min,51.5968min及103.2624min三个色谱峰保留时间和峰面积为参照计算,精密度、稳定性和重现性的相对保留时间及相对峰面积rsd值均符合2015版《中国药典》关于指纹图谱方法建立方法学验证的相关规定。
4.不同批次苏黄止咳胶囊指纹图谱建立
根据按1项下色谱条件及2项下方法制备供试品溶液,建立16批次苏黄止咳胶囊指纹图谱,并采用国家药典委员会的“中药色谱指纹图谱相似度评价系统软件(2012版)”进行评价,设定样品s8图谱为参照图谱,时间窗为0.10min,以平均数法进行相似度计算,生成苏黄止咳胶囊对照指纹图谱,见附图5。通过与对照品比对,共指认了11个色谱峰,对照品保留时间,见表7。16批次苏黄止咳胶囊有22个共有峰,见附图6,占各批次样品中总峰面积的40%。各批次样品相似度值,见表8。
表711个已指认共有峰成分保留时间(min)
表816批次苏黄止咳胶囊样品相似度
相似度分析结果表明,16个不同批次苏黄止咳胶囊相似度均大于0.9,表明苏黄止咳胶囊在两年保质期内质量稳定。
实施例2
苏黄止咳胶囊及其主要成分抗炎活性研究
1.材料与方法
1.1材料
小鼠单核巨噬细胞系raw264.7(上海中国科学院细胞库);dmem培养基(美国biologicalindustries公司,010521acs);胎牛血清(美国biologicalindustries公司,040011acs);脂多糖(美国sigma-aldrich公司,l2630)。synergyh1多功能微孔板检测仪(美国biotek公司)。
1.2方法
1.2.1细胞培养
raw246.7细胞培养在dmem培养液(含10%fbs和1%青霉素-链霉素)中,置于37℃、5%co2培养箱中培养,根据生长情况更换培养液,用培养液轻轻吹落细胞,收集细胞悬浮液,在1000r/min下离心2min后弃去上清液,再加入3ml细胞培养液重悬细胞,按1∶3的体积比接种至新的培养皿中培养。
1.2.2脂多糖诱导raw246.7细胞炎症模型的建立及筛选
取对数生长期细胞按照2×104个/孔的数目接种到96孔板中,置于在37℃、5%的co2培养箱中培养24h。观察细胞状态,设置不同实验组:(1)空白对照组,不做任何处理;(2)lps处理组,用浓度为1μg·ml-1的lps持续刺激细胞12h;(3)药物+lps处理组,苏黄止咳胶囊提取物、地塞米松、不同浓度牛蒡苷元、五味子醇甲、白花前胡甲素、麻黄碱及伪麻黄碱预处理raw264.7细胞24h后,加入1μg·ml-1的lps继续处理24h后收集细胞培养上清液。各组分别设置6个复孔。取100μl细胞上清液加入100μlgriess试剂,室温下避光反应15min,用酶标仪在540nm下测定吸光度值。计算抑制率,求半数抑制浓度(ic50)值。
抑制率(%)=(a1-a2)/(a1-a0)×100
其中a1为模型组吸光度值,a2为给药组吸光度值,a0为空白组吸光度值。
1.3结果
各受试药物的活性测试结果表明,白花前胡甲素的ic50值为21.35±5.87μm,与阳性对照组地塞米松ic50在值19.07±5.65μm比较没有显著性差异,活性相当,而苏黄止咳胶囊提取物、牛蒡苷元、伪麻黄碱和五味子醇甲则活性较弱,但仍可在一定程度抑制lps诱导raw264.7炎症细胞产生的no生成活性。
表9各成分筛选no生成活性的ic50
对比例
1.流动相对比考察
流动相组成对样品色谱行为影响较大,因此在保证其他色谱条件一致情况下,通过改变不同流动相组成来优化较好的流动相。本申请主要考察实验室常用有机色谱溶剂(甲醇、乙腈)对样品分离情况的影响。结果,见附图7。其中:s1为乙腈-水,s2为乙腈-0.05%甲酸水,s3为甲醇-0.05%甲酸水。结果表明,当有机相为乙腈时样品分离效果较甲醇好,且在较短时间内可完成分离。
流动相洗脱梯度直接影响样品色谱峰的分离情况及样品分析时间,因此在控制样品制备方法和其他色谱条件一致情况下考察不同洗脱梯度对苏黄止咳胶囊提取物分离效果。其中流动相洗脱梯度分别为:乙腈(a)-0.05%甲酸水(b),流动相洗脱梯度:(1)0~5min,5%(a);5~25min,5~26%(a);25~45min,26~40%(a);45~55min,40~54%(a);55~85min,54~66%(a);85~95min,66~71%(a);95~105min,71~86%(a);105~115min,86~92%(a);115~135min,92~95%(a);135~140min,95~100%(a)。(2)0~15min,4%(a);15~40min,4~15%(a);40~90min,15~35%(a);90~110min,35~50%(a);110~130min,50~60%(a);130~140min,60~80%(a);140~145min,80~95%(a)。(3)0~15min,4%(a);15~40min,4~15%(a);40~90min,15~35%(a);90~110min,35~50%(a);110~130min,50~60%(a);130~140min,60~90%;140~145min,90~95%(a)。结果,见附图8。通过优化不同洗脱梯度,结果当按洗脱梯度(3)运行样品时所得图谱基线平稳,且色谱峰得到较好分离。
流动相中加入酸可调节流动相ph值,从而可改善峰型。本实验考察实验室流动相加入常用的各种酸,即乙酸(体积分数为0.02%和0.05%)、甲酸(体积分数为0.02%和0.05%)和磷酸(体积分数为0.02%和0.05%)对苏黄止咳胶囊样品分离情况的影响。结果,见附图9。如图所示,当流动相中不加酸时(s1)大极性部分色谱峰分离情况较差,而水相中加入不同浓度乙酸时色谱峰分离情况(s2和s3)不如加入甲酸好,水相中加入0.05%甲酸水时(s5)与0.02%磷酸时(s6)色谱峰分离的较好,但磷酸为无机酸色谱体系容易出现盐析现象。因此,选择水相中加入0.05体积%甲酸。
2.色谱柱对比考察
不同产家色谱柱由于填料组成上的差异对样品分离影响较大,因此本部分实验拟通过优化不同产家及型号色谱柱,优选较优色谱柱。其中所优化色谱柱分别为a:agilent(zorbaxsb-c18,4.6×250mm,5μm,pn:880957-902);b:agilent(zorbax,sb-c18,4.6×150mm,5μm,pn:883975-902);c:waters(
3.检测波长对比考察
苏黄止咳胶囊中成分组成复杂,包含生物碱、木脂素、香豆素、黄酮、三萜类等成分,而不同成分最大吸收波长各异。本实验考察230、248、254、270、300、330nm六个不同波长下对样品检测情况的影响。结果,见附图11。结果如图所示,当检测波长为230nm(s1)时基线漂移较严重,而当检测波长超过270nm(s4)时,小极性部分色谱峰响应较低。选择检测波长254nm(s3)色谱峰信息较多。
4.流速对比考察
不同流速会影响色谱峰出峰时间,进而影响样品分离情况。本实验通过考察不同流速(0.6、0.8、1.0、1.2、1.4ml·min-1)对苏黄止咳胶囊样品分离情况的影响,从而优化出较好色谱条件。结果,见附图12。结果表明,当流速为1ml·min-1(s3)时样品分离时间较短,且色谱峰分离的较好。
5.柱温对比考察
柱温不仅可以影响样品分离情况,亦可影响样品运行时间。本实验考察不同柱温(20、26、28、30℃)对苏黄止咳胶囊样品分离情况的影响。结果,见附图13。结果如图所示,当选择柱温为28℃(s3)时样品分离情况较好。
虽然本申请所揭露的实施方式如上,但所述的内容仅为便于理解本申请而采用的实施方式,并非用以限定本申请。任何本申请所属领域内的技术人员,在不脱离本申请所揭露的精神和范围的前提下,可以在实施的形式及细节上进行任何的修改与变化,但本申请的专利保护范围,仍须以所附的权利要求书所界定的范围为准。
- 还没有人留言评论。精彩留言会获得点赞!