枸橼酸托法替布中氰基乙酸乙酯的分析方法与流程
1.本发明属于医药检测分析技术领域,特别涉及一种枸橼酸托法替布中氰基乙酸乙酯的分析方法。
背景技术:
2.枸橼酸托法替布,化学名称为(3r,4r)
‑4‑
甲基
‑3‑
(甲基
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基氨基)
‑
β
‑
氧代
‑1‑
哌啶丙腈枸橼酸盐,化学结构式如下图所示:枸橼酸托法替布是第一个被批准用于人类自身免疫性疾病的小分子非受体型酪氨酸蛋白激酶抑制剂,主要用于甲氨蝶呤疗效不足或对其无法耐受的中度至重度活动性类风湿关节炎(ra)成年患者,可与甲氨蝶呤或其他非生物改善病情抗风湿药(dmard)联合使用。
[0003]
杂质研究为药物研发过程中的重要部分,与药品的安全性和有效性直接相关。枸橼酸托法替布在合成中杂质的主要来源是起始原料引入的杂质和工艺杂质,氰基乙酸乙酯为枸橼酸托法替布的起始物料之一,该物质无生色基团,紫外吸收很弱,而且沸点较高,不易气化,检测难度较大,为保证原料药质量,需严格控制该杂质含量。因此,开发一种操作简单、准确度高、回收率稳定的检测托法替布中氰基乙酸乙酯的方法非常有必要。
技术实现要素:
[0004]
本发明的目的在于提供一种操作简单、回收率稳定、准确度高的枸橼酸托法替布中氰基乙酸乙酯的分析方法。
[0005]
根据本发明的一个方面,提供一种枸橼酸托法替布中氰基乙酸乙酯的分析方法,所述分析方法采用气相色谱法,色谱柱为以6%氰丙基苯基
‑
94%二甲基聚硅氧烷为固定液的毛细管柱,稀释剂为乙腈
‑
稀酸溶液,所述稀酸溶液的质量浓度为0.1~0.2%。
[0006]
进一步地,所述稀酸溶液选自磷酸溶液、甲酸溶液、醋酸溶液或三氟乙酸溶液,所述乙腈与所述稀酸溶液的体积比为40~70:60~30。
[0007]
进一步地,所述稀酸溶液选自0.1%磷酸溶液。
[0008]
进一步地,所述的枸橼酸托法替布中氰基乙酸乙酯的分析方法,包括以下步骤:1)取枸橼酸托法替布待测品用稀释剂配制成供试品溶液,2)取氰基乙酸乙酯对照品用稀释剂配制成对照品溶液,
3)取所述供试品溶液、对照品溶液,注入气相色谱仪,记录色谱图。
[0009]
进一步地,步骤3)中,气相色谱检测条件为:检测器温度:250℃;进样口温度:250℃;柱流量:1.3~1.8ml/min;分流比:1~5∶1;进样量为2~5μl;柱温:起始温度100
±
5℃,维持5~10分钟,以每分钟10~20℃的速率上升到200~220℃,维持5~10分钟。
[0010]
进一步地,所述对照品溶液中氰基乙酸乙酯的浓度为1~20μg/ml,优选4~20μg/ml。
[0011]
进一步地,所述供试品溶液的浓度为3~10mg/ml。
[0012]
进一步地,所述氰基乙酸乙酯的检测限浓度为0.2732μg/ml;定量限浓度为1.0927μg/ml。
[0013]
进一步地,氰基乙酸乙酯溶液浓度在0.8802μg/ml~23.4720μg/ml范围与峰面积具有线性。
[0014]
本发明的有益效果:本发明的枸橼酸托法替布中氰基乙酸乙酯的分析方法,样品配制简单,无需用溶剂先溶解再稀释,可以直接用稀释剂溶解稀释定容,一步到位,提高实验效率,并且本发明的分析方法样品回收率稳定,在50%~150%的浓度范围,氰基乙酸乙酯的回收率在94.39%~102.38%之间,平均回收率为97.97%,rsd为2.78%,准确度高。
附图说明
[0015]
图1
‑
1为实施例1的对照品溶液的色谱图;图1
‑
2为实施例1的供试品溶液的色谱图;图2
‑
1为实施例2的对照品溶液的色谱图;图2
‑
2为实施例2的供试品溶液的色谱图;图3
‑
1为实施例3的对照品溶液的色谱图;图3
‑
2为实施例3的供试品溶液的色谱图;图4
‑
1为实施例4的对照品溶液的色谱图;图4
‑
2为实施例4的供试品溶液的色谱图。
具体实施方式
[0016]
下面通过具体实施例对本技术作进一步详细说明。以下实施例仅对本技术进行进一步说明,不应理解为对本技术的限制。
[0017]
本实施例提供一种枸橼酸托法替布中氰基乙酸乙酯的分析方法,采用气相色谱法直接进样,采用外标法进行定量,色谱柱选自以6%氰丙基苯基
‑
94%二甲基聚硅氧烷为固定液的毛细管柱,稀释剂选自乙腈
‑
稀酸溶液,稀酸溶液的质量浓度为0.1~0.2%。乙腈中加入一定量的稀酸溶液,增大了枸橼酸托法替布的溶解度,可以直接用稀释剂溶解枸橼酸托法替布并稀释定容制成供试品溶液,无需像现有技术中那样先用溶剂溶解再用稀释剂定容,方便实验人员操作,提高实验效率。乙腈与稀酸溶液的体积比优选为40~70:60~30。稀酸溶液优选为磷酸溶液、甲酸溶液、醋酸溶液或三氟乙酸溶液,更优选为磷酸溶液。本发明的稀释剂采用乙腈
‑
稀酸溶液,不仅可以增加枸橼酸托法替布的溶解度,配制样品时溶解稀释定容一步到位,并且本发明的分析方法样品回收率稳定,在50%~150%的浓度范围,氰基乙酸乙酯的回收率在94.39%~102.38%之间,平均回收率为97.97%,rsd为2.78%,准确度高,专
属性强,空白溶液对氰基乙酸乙酯的检测无干扰,混合溶液中,氰基乙酸乙酯与相邻峰之间的分离度均大于1.5。
[0018]
本实施例的枸橼酸托法替布中氰基乙酸乙酯的分析方法,具体包括以下步骤:1)取枸橼酸托法替布待测品用稀释剂配制成供试品溶液,供试品溶液的浓度为3~10mg/ml。
[0019]
2)取氰基乙酸乙酯对照品用稀释剂配制成对照品溶液,对照品溶液中氰基乙酸乙酯的浓度为1~20μg/ml,优选4~20μg/ml。
[0020]
3)取供试品溶液、对照品溶液,注入气相色谱仪,记录色谱图。
[0021]
气相色谱检测条件为:检测器温度:250℃;进样口温度:250℃;柱流量:1.3~1.8ml/min;分流比:1~5∶1;进样量为2~5μl;柱温:起始温度100
±
5℃,维持5~10分钟,以每分钟10~20℃的速率上升到200~220℃,维持5~10分钟。
[0022]
本实施例所用分析仪器为agilent 7890b气相色谱仪,磷酸、三氟乙酸为ar级,乙腈为hplc级,对照品氰基乙酸乙酯为ar级,枸橼酸托法替布为自制。
[0023]
实施例1稀释剂溶液:乙腈
‑
0.1%磷酸水溶液(50∶50)。
[0024]
供试品溶液配制:取枸橼酸托法替布0.1g,精密称定,置10ml量瓶中,加稀释剂溶液并稀释至刻度,摇匀,即得。
[0025]
对照品溶液:取氰基乙酸乙酯对照品适量,加稀释剂制成每1ml约含氰基乙酸乙酯10μg的溶液。
[0026]
取上述供试品溶液、对照品溶液各5μl,注入色谱仪,记录色谱图。见图1
‑
1和图1
‑
2,图1
‑
1为对照品溶液的色谱图;图1
‑
2为供试品溶液的色谱图。由图可知本实施例的分析方法,对照品氰基乙酸乙酯能够有效检出,供试品里的峰不会干扰到对照品。
[0027]
色谱条件如下:色谱柱:6%氰丙基苯基
‑
94%二甲基聚硅氧烷为固定液的毛细管柱,本实施例中色谱柱规格/型号为agilent db
‑
624 ui(30m
×
0.32mm
×
1.8μm),fid检测器;检测器温度:250℃;进样口温度:250℃;进样量:5μl;柱流量:1.3ml/min;分流比:3∶1;溶剂:乙腈
‑
0.1%磷酸水溶液(50∶50);柱温:起始温度100℃,维持6分钟,以每分钟15℃的速率上升到220℃,维持8分钟。
[0028]
实施例2稀释剂溶液:乙腈
‑
0.1%三氟乙酸水溶液(50∶50)。
[0029]
供试品溶液配制:取枸橼酸托法替布0.1g,精密称定,置10ml量瓶中,加稀释剂溶液并稀释至刻度,摇匀,即得。
[0030]
对照品溶液:取氰基乙酸乙酯对照品适量,加稀释剂制成每1ml约含氰基乙酸乙酯10μg的溶液。
[0031]
取上述供试品溶液、对照品溶液各5μl,注入色谱仪,记录色谱图。见图2
‑
1和图2
‑
2,图2
‑
1为对照品溶液的色谱图;图2
‑
2为供试品溶液的色谱图。由图可知本实施例的分析方法,对照品氰基乙酸乙酯能够有效检出,供试品里的峰不会干扰到对照品。
[0032]
色谱条件如下:色谱柱:6%氰丙基苯基
‑
94%二甲基聚硅氧烷为固定液的毛细管柱,本实施例中
色谱柱规格/型号为agilent db
‑
624 ui(30m
×
0.32mm
×
1.8μm),fid检测器;检测器温度:250℃;进样口温度:250℃;进样量:5μl;柱流量:1.8ml/min;分流比:3∶1;溶剂:乙腈
‑
0.1%三氟乙酸水溶液(50∶50);柱温:起始温度100℃,维持5分钟,以每分钟15℃的速率上升到220℃,维持8分钟。
[0033]
实施例3稀释剂溶液:乙腈
‑
0.1%磷酸水溶液(40∶60)。
[0034]
供试品溶液配制:取枸橼酸托法替布0.1g,精密称定,置10ml量瓶中,加稀释剂溶液并稀释至刻度,摇匀,即得。
[0035]
对照品溶液:取氰基乙酸乙酯对照品适量,加稀释剂制成每1ml约含氰基乙酸乙酯20μg的溶液。
[0036]
取上述供试品溶液、对照品溶液各5μl,注入色谱仪,记录色谱图。见图3
‑
1和图3
‑
2,图3
‑
1为对照品溶液的色谱图;图3
‑
2为供试品溶液的色谱图。由图可知本实施例的分析方法,对照品氰基乙酸乙酯能够有效检出,供试品里的峰不会干扰到对照品。
[0037]
色谱条件如下:色谱柱:6%氰丙基苯基
‑
94%二甲基聚硅氧烷为固定液的毛细管柱,本实施例中色谱柱规格/型号为agilent db
‑
624 ui(30m
×
0.32mm
×
1.8μm),fid检测器;检测器温度:250℃;进样口温度:250℃;进样量:5μl;柱流量:1.5ml/min;分流比:3∶1;溶剂:乙腈
‑
0.1%磷酸水溶液(50∶50);柱温:起始温度100℃,维持5分钟,以每分钟10℃的速率上升到220℃,维持8分钟。
[0038]
实施例4稀释剂溶液:乙腈
‑
0.2%磷酸水溶液(60∶40)。
[0039]
供试品溶液配制:取枸橼酸托法替布0.1g,精密称定,置10ml量瓶中,加稀释剂溶液并稀释至刻度,摇匀,即得。
[0040]
对照品溶液:取氰基乙酸乙酯对照品适量,加稀释剂制成每1ml约含氰基乙酸乙酯4μg的溶液。
[0041]
取上述供试品溶液、对照品溶液各5μl,注入色谱仪,记录色谱图。见图4
‑
1和图4
‑
2,图4
‑
1为对照品溶液的色谱图;图4
‑
2为供试品溶液的色谱图。由图可知本实施例的分析方法,对照品氰基乙酸乙酯能够有效检出,供试品里的峰不会干扰到对照品。
[0042]
色谱条件如下:色谱柱:6%氰丙基苯基
‑
94%二甲基聚硅氧烷为固定液的毛细管柱,本实施例中色谱柱规格/型号为agilent db
‑
624 ui(30m
×
0.32mm
×
1.8μm),fid检测器;检测器温度:250℃;进样口温度:250℃;进样量:5μl;柱流量:1.5ml/min;分流比:3∶1;溶剂:乙腈
‑
0.2%磷酸水溶液(50∶50);柱温:起始温度100℃,维持5分钟,以每分钟15℃的速率上升到220℃,维持8分钟。
[0043]
方法学验证(1)专属性空白溶液(稀释剂):乙腈
‑
0.1%磷酸水溶液(50∶50)各溶液的定位溶液:取甲醇约300mg、乙醇约500mg、丙酮约500mg、叔丁基甲基醚约500mg、二甲基亚砜约500mg、二氯甲烷约60mg、乙酸乙酯约500mg、四氢呋喃约72mg、氰乙酸
约100mg、甲苯约89mg和正丁醇约500mg,精密称定,分别置100ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。
[0044]
苯定位溶液:取苯约20mg,精密称定,置100ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀;取上述溶液1ml置100ml量瓶中,用稀释剂稀释至刻度,摇匀,即得。
[0045]
对照品贮备液:取氰基乙酸乙酯约20mg,精密称定,置20ml量瓶中,用稀释剂溶解并稀释至刻度,摇匀,即得。(1mg/ml)对照品溶液:取对照品贮备液1ml,置100ml量瓶中,用稀释剂稀释至刻度,摇匀,即得。(10μg/ml)供试品溶液:取本品约0.1g,精密称定,置10ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。(10mg/ml)混合溶液:取本品约0.2g,精密称定,置20ml量瓶中,取各溶液的定位溶液各1ml,置同一20ml的量瓶中,加稀释剂稀释至刻度,摇匀,即得。
[0046]
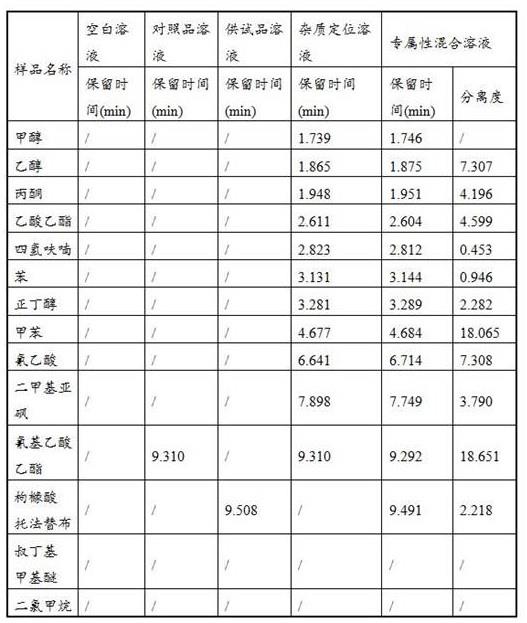
取上述空白溶液、对照品溶液、供试品溶液、混合溶液、各溶剂定位溶液进样分析,记录色谱图,结果见下表。
[0047]
表1专属性结果表
结果表明,空白溶剂对氰基乙酸乙酯的检测无干扰,各溶液对氰基乙酸乙酯的检测无干扰,混合溶液中氰基乙酸乙酯与相邻溶剂间的分离度符合要求。
[0048]
(2)检测限和定量限精密量取对照品溶液适量,逐级稀释,使s/n约为3时,即为检测限;使s/n约为10时,即为定量限。分别取各溶液进行分析,结果见下表。
[0049]
表2检测限与定量限结果
结果表明:该方法中氰基乙酸乙酯的检测限浓度为0.2732μg/ml;定量限浓度为1.0927μg/ml。
[0050]
(3)线性取定量限溶液、40%、60%、80%、100%、120%、160%、200%线性溶液进样分析(各3针),记录色谱图。以浓度为横坐标(c)、平均峰面积为纵坐标(a)进行线性回归,并求出线性方程。实验结果见下表。
[0051]
表3线性结果表 从上表可知,氰基乙酸乙酯溶液浓度在0.8802μg/ml~23.4720μg/ml范围,浓度与峰面积线性方程为y=32798.7461x
‑
10920.8369,相关系数r=0.9998,说明线性关系良好。
[0052]
(4)溶液稳定性取对照品+供试品溶液在0h、2h、4h、6h、8h、12h进行分析,结果见下表。
[0053]
表4溶液稳定性结果
ꢀ
结果表明,室温条件下,对照品+供试品溶液在12h内溶液稳定。
[0054]
(5)精密度
①
进样精密度取对照品溶液进样分析,记录色谱图,计算各峰面积rsd,结果见下表。
[0055]
表5进样精密度结果表 从上表可知,对照品溶液重复进样6次所得保留时间rsd为0.01%,峰面积的rsd为0.34%,说明仪器的精密度良好。
[0056]
②
重复性取对照品溶液和供试品溶液(平行配制6份)进样分析,记录色谱图,按外标法计算结果,结果见下表。
[0057]
表6重复性测试结果表 结果表明,6份样品中氰基乙酸乙酯均未检出,说明该方法的重复性较好。
[0058]
③
中间精密度不同实验人员在不同时间分别配制对照品溶液和6份供试品溶液,进样分析,记录色谱图,按外标法计算结果,结果见下表。
[0059]
表7中间精密度测试结果表 结果表明,12份供试品中氰基乙酸乙酯均未检出。
[0060]
(6)准确度取供试品溶液、对照品溶液、各浓度水平(50%、100%、150%)溶液进样分析,记录色谱图,结果见下表。
[0061]
表8回收率结果表从上表可知,在50%~150%的浓度范围,氰基乙酸乙酯的回收率在94.39%~102.38%之间,平均回收率为97.97%,rsd为2.78%,说明该方法准确度较好。
[0062]
(7)耐用性通过改变柱流量、初始柱温来评估测定条件参数有微小的变动时,测定结果不受影响的承受程度。分别取空白溶液、对照品溶液和供试品溶液进行分析,结果见下表。
[0063]
表9耐用性各色谱条件表表10不同色谱条件耐用性测试结果结论:分别调节柱流量(1.3ml/min~1.8ml/min)、起始柱温(
±
5℃),进行检测,供试品中氰基乙酸乙酯均未检出。说明测定条件参数有微小的变动时,测定结果基本不受影响,即方法耐用性较好。
[0064]
以上内容是结合具体的实施方式对本技术所作的进一步详细说明,不能认定本技术的具体实施只局限于这些说明。对于本技术所属技术领域的普通技术人员来说,在不脱离本技术构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本技术的保护
范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1