一种基于分层混合模型的信号肽及其切割位点的预测方法与流程
本发明涉及一种基于分层混合模型的信号肽及其切割位点的预测方法,是利用已知的蛋白质序列来预测该蛋白质是否包含n端信号肽,并预测其切割位点,特别是一种融合氨基酸残基和功能结构域,融合统计性信任分数和序列相似性分数,并自顶而下分层预测信号肽及其切割位点的算法。
背景技术:
1979年,g.blobel和d.sabatini基于实验观察首次提出了信号假说(signalhypothesis)。g.blobel和d.sabatini认为在分泌蛋白质序列的n端有一段起信号引导作用的氨基酸片段,该片段可以引导蛋白质在各个膜间进行转移,并将蛋白质运输到目的位置。他们把这段起信号引导作用的氨基酸片段称为信号肽。在1999年,g.blobel凭借这项成就获得了诺贝尔生理学或医学奖。
经过多年的研究对信号肽的研究,人们对信号肽的认识进一步加深。信号肽是一段出现在蛋白质序列n端的氨基酸短序列,它在真核生物和原核生物中引导蛋白质在细胞中的转移和分泌。几乎所有的分泌蛋白质和许多跨膜蛋白质氨基酸序列的n端都包含信号肽。作为一个“邮编”,信号肽引导新生蛋白质运输到细胞内外的正确位置。如果改变了新生蛋白n端的信号肽,那么蛋白质可能会被运输到错误的细胞位置,这将导致各种奇怪的疾病。因此,信号肽的知识对揭示了一些复杂遗传性疾病的机制是非常有用的。此外根据信号肽的知识,科学家可以自己期望的方式对氨基酸序列进行重新编程,这可以用于未来的细胞和基因治疗中,并同时为开发新药物提供了思路。事实上,药物科学家通过基因修饰蛋白质对应信号肽,例如,科学家可以通过向所需蛋白质添加特异性标签(信号肽),可以标记并引导它们的排泄,使得它们更容易被获取。现在,信号肽已成为寻找新药物,对细胞进行重新编程和进行基因治疗的关键工具。此外,信号肽预测还可以为其他相关领域的预测提供重要的参考信息,如在亚细胞定位研究中,信号肽的信息可以直接提供蛋白质的亚细胞位置信息。
在过去的二十年里,科学一直在信号肽预测及其位点分析领域孜孜不倦的努力,并取得了出色的研究成果。到目前为止,领域内不仅发布了多种信号肽分析的方法,而且也有许多信号肽分析软件发布并被广泛使用。领域内的信号肽分析方法逐步提升了信号肽分析的精度,并尝试从不同的角度分析认识信号肽。统计常用的信号肽分析软件,根据其进行信号肽分析时应用的预测模型,我们可以将它们大致分成三类:基于生成模型的信号肽预测方法,基于判别模型的信号肽预测方法和基于序列比对的信号肽预测方法。基于判别模型的信号肽分析方法通常是应用统计分析分类器(如神将网络,支持向量机等等)进行信号肽分析。这种方法的优点是比较灵活,适合处理任意长度的蛋白质序列,缺点是由于每个氨基酸残基被独立地编码表示,各氨基酸残基间的相互关联就被忽略。此外,在基于判别模型的信号肽分析方法进行切割位点判定时,容易在真实切割位点左右两边陷入局部最优陷阱。基于生成模型的信号肽分析方法考虑了信号肽结构上的三个功能区(n区,h区,c区)信息,此类方法通过统计信号肽各区域间的相关性,并通过隐马尔可夫模型(hiddenmarkovmodel,hmm)等算法进行建模。基于生成模型的信号肽分析方法优点是在生物学和化学等层面上有更加直观的可解释性,并且探索信号肽功能域间相关性,这将有助于更深刻的认识这三个功能区域的功能和结构;缺点是不擅长处理信号肽具有较长氨基酸序列的情况。基于序列比对的信号肽分析方法是基于信号肽的进化保守性和知识迁移等思想构建的,在这类信号肽分析方法中,首先需要从公共数据库中提取信号肽序列组成包含注释信息的数据集,然后将查询序列与数据集中的信号肽序列进行序列比对。这类信号肽分析方法的优点是在生物学和物理学上有较强的可解释性,该类模型相比其他两类模型更为灵活,可以通过动态的更新模型使用的数据集来更新发布的信号肽分析模型,不需要在新的数据集上重新训练模型。
对信号肽预测来说,基于氨基酸残基特征分类器能很好地区分信号肽和非分泌蛋白质。但是信号肽和n端跨膜螺旋片段在结构上都有一段呈疏水性的氨基酸片段,所以它们氨基酸残基特征有较高的相似性。因此仅凭氨基酸残基特征,分类器难以正确识别信号肽和跨膜螺旋,以至于信号肽预测的假阳性过高。信号肽切割位点预测一直以来都是一个难点,原因可能是信号肽切割位点具有复杂的模式,信号肽长度分布较为分散。
技术实现要素:
本发明的目的在于针对现有技术中的不足,提供本发明提出一种基于分层混合模型的信号肽及其切割位点预测算法,该算法在信号肽预测领域第一次引入功能性结构域信息来降低n端跨膜螺旋的假阳性比率,融合氨基酸残基和功能性结构域信息后信号肽预测性能显著提高。在信号肽切割位点预测方面,该算法通过统计规则筛选高质量切割位点候选集,然后通过序列比对计算候选切割位点的序列相似性分数,通过融合统计性信任分数和序列相似性分数预测最终的信号肽切割位点。本发明在信号肽预测方面有较低的假阳性,在信号肽切割位点预测方面有较高的灵敏度。
本发明所解决的技术问题可以采用以下技术方案来实现:
首先在第一层应用基于氨基酸残基特征的svm分类器识别蛋白质序列是否含有n端疏水性片段;然后在第二层应用基于氨基酸残基特征和功能性结构域特征对应的朴素贝叶斯和svm分类器识别疏水性片段是信号肽还是n端跨膜螺旋;最后在第三层,先根据统计学习规则筛选候选切割位点并计算统计性信任分数,然后再通过needleman-wunsch序列比对算法计算候选信号肽序列的相似性分数,对统计性信任分数和序列相似性分数积分确定预测的信号肽切割位点。
其具体步骤是:
第一步:利用蛋白质氨基酸序列提取pssm矩阵信息,二级结构信息,可溶性信息和氨基酸物理化学信息,使用mrmr进行特征提取,生成氨基酸序列特征。
第二步:从cdd(conserveddomaindatabase)数据库中提取蛋白质序列的功能性结构域信息,并根据cdd提供的聚类信息将功能结构信息映射成超家族,并根据超家族出现的频率生成频繁功能结构特征。
第三步:根据蛋白质序列的两类特征分别训练svm分类器和朴素贝叶斯分类器用于信号肽预测。
第四步:对于预测为信号肽的蛋白质序列,使用不对称滑动窗口[-13,+2]在蛋白质序列上生成氨基酸序列片段。
第五步:根据信号肽的{-3,-1,+1}规则,在生成的氨基酸序列片段中筛选候选的信号肽切割位点,并计算每个候选切割位点对应的统计性信任分数。
第六步:由候选的信号肽切割位点生成对应的信号肽候选序列,并将该序列和含有注释信息的信号肽序列应用needleman-wunsch算法做序列比对,并计算每条信号肽候选序列对应的序列相似性分数和比通过序列对得到的信号肽切割位点。
第七步:对于每个候选切割位点对应的统计性信任分数和序列相似性分数进行积分,计算最终预测的信号肽切割位点。
与现有技术相比,本发明的有益效果如下:
(1)模型融合氨基酸残基和序列功能结构域信息,显著降低信号肽预测的假阳性;
(2)模型融合统计性分数和序列相似性分数,显著提高了信号肽切割位点预测的灵敏度;
(3)自顶而下分层预测信号肽及其切割位点的模型,显著提高了信号肽及其切割位点预测的性能。
附图说明
图1是本发明所述的基于分层混合模型的信号肽及其切割位点预测方法的示意图。
图2是本发明所述的基于分层混合模型的信号肽及其切割位点预测方法的输出结果示意图。
具体实施方式
为使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施方式,进一步阐述本发明。
参见图1,本发明所述的基于分层混合模型的信号肽及其切割位点预测方法,首先在第一层应用基于氨基酸残基特征的svm分类器识别蛋白质序列是否含有n端疏水性片段;然后在第二层应用基于氨基酸残基特征和功能性结构域特征对应的朴素贝叶斯和svm分类器识别疏水性片段是信号肽还是n端跨膜螺旋;最后在第三层,先根据统计学习规则筛选候选切割位点并计算统计性信任分数,然后再通过needleman-wunsch序列比对算法计算候选信号肽序列的相似性分数,对统计性信任分数和序列相似性分数积分确定预测的信号肽切割位点。
下面具体进行阐述:
第一步:根据蛋白质序列信息提取氨基酸残基对应的进化信息(pssm),结构信息(ss)和物理化学信息(pi),这样蛋白质序列中的每个残基可以对应的一个30维向量表示:
rf=[pssm(20),ss(5),pi(5)](1)
第二步:取蛋白质序列n端l个氨基酸对应的特征,然后应用mrmr算法进行特征选择生成优化后的特征记为srf;
第三步:应用基于srf的svm分类器判断蛋白质序列是否包含疏水性片段(信号肽和n端跨膜螺旋),若不含有分析结束;
第四步:使用rps-blast软件检索cdd数据库获取蛋白质序列的功能性结构域信息,并根据结构域id和结构域超家族的映射关系生成能性结构域特征(fdf);
fdfcdd=[δ1δ2…δi…δm](2)
第五步:应用基于fdf的朴素贝叶斯分类器判断疏水性氨基酸片段是信号肽还是n端跨膜螺旋,对没有fdf的蛋白质采用基于srf特征的svm分类器进行预测;
第六步:对预测为包含信号肽的蛋白质序列进行切割位点预测,使用[-13,+2]滑动窗口生成氨基酸片段,并根于{-3,-1,+1}规则和统计学习算法筛选候选的切割位点集;
γ={rk},ifθk>φt(5)
第七步:根据筛选出的候选切割位点生成假设的序列片段,并将片段和含有注释信息的蛋白质序列做序列比对,计算相似性分数;
第八步:对每个候选切割位点对应的统计性信任分数和序列相似性分数进行积分,选取最大值对应的候选切割位点为最后预测的信号肽切割位点。
实施例
现有一个输入序列,数据如下:
>querysequence|signal125
miksnritacalaalfagasfsasawwggpgygnglwdnmgdmfgdgygdfnmsm
ggggrgygrgygrgngygygapygygapygygapygygapygygapygampyga
mppqmpaapaqpqaapsr
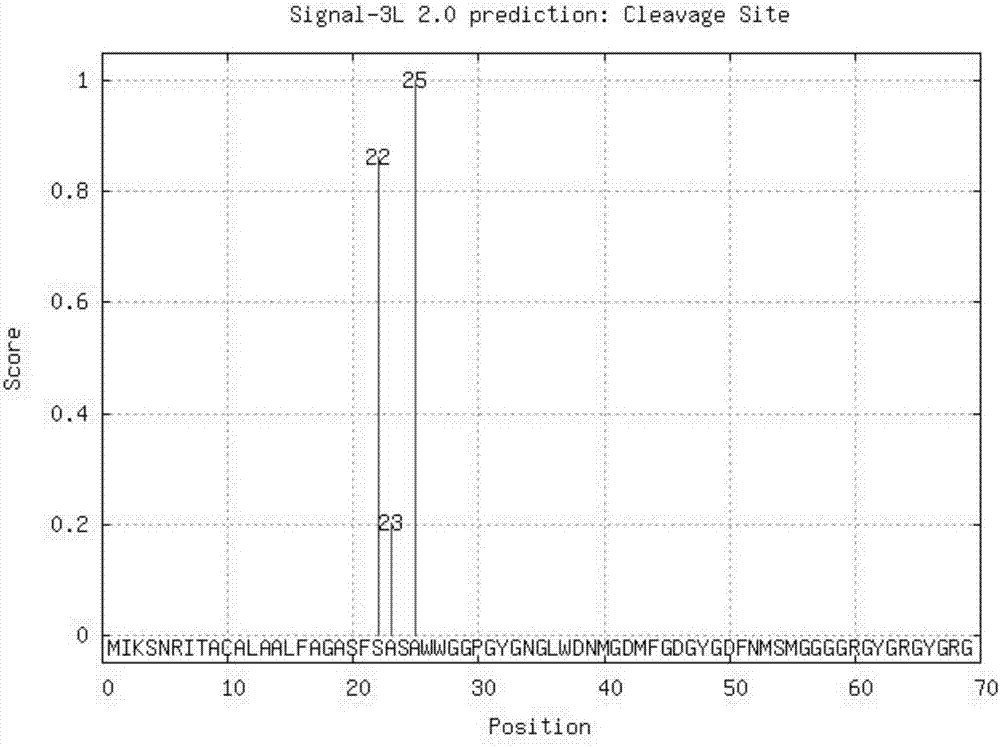
此为一个待测序列,使用本发明方法的软件输出结果如图2所示:
accordingtosignal-3l2.0engine,thepredictedsignalpeptideis:1-25
miksnritacalaalfagasfsasawwggpgygnglwdnmgdmfgdgygdfnmsm
ggggrgygrgygrgn
theprotentialcleavagesitesandthecreditscores
从结果可以看出,本方法精确并且直观的预测了信号肽及其切割位点。
以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!