一种蛋白质折叠类型分类方法与流程
本发明涉及一种蛋白质折叠类型的自动化分类方法。
背景技术:
蛋白质三级结构复杂而不规则,但其所对应的蛋白质折叠类型却只有数百到数千种,蛋白质折叠类型分类是蛋白质折叠类型首先需要解决的基本问题。scop数据库是应用最广泛的结构分类数据库,为层状结构,包括蛋白质结构类、折叠类型、超家族、家族等不同层次,与蛋白质折叠类型对应的是fold层次,它是在超家族的基础上,按照二级结构及其空间分布及拓扑连接,根据专家经验人工完成折叠类型的指认。2013年,在scop已有分类的基础上,scop数据库建立。尽管scop中部分蛋白质样本通过序列比对可自动获得分类结果,但所用自动分类结果与手动分类结果并不相同。新发布的astral现在依然使用scop中的手动分类结果。最近7年,scop数据中折叠层所包含的折叠类型总数基本保持在1393种左右,四种主要结构类包含的折叠类型总数保持在1000种左右,折叠类型总数基本稳定。因此,对已有scop的人工分类结果进行数据挖掘、建立蛋白质折叠类型分类方法,实现蛋白质折叠类型的自动分类,是迫切需要解决的问题。
技术实现要素:
为了克服上述缺陷,本发明提供一种基于统一原理的蛋白质折叠类型分类方法,从而实现蛋白质折叠类型的自动化分类。
为了实现上述目的,本发明采用的技术方案是:
一种蛋白质折叠类型分类方法包括以下步骤:
步骤1、构建模板数据库;
步骤2、基于模板数据库,将任意待测蛋白样本与模板数据中的所有模板进行tm-align比对,计算tm-score值,所述tm-score取值最大的模板所在的折叠类型即为待测蛋白样本所属折叠类型。
作为优选,所述模板数据库包括家族模板数据库与折叠类型模板数据库。
作为优选,所述家族模板数据库构建为:对家族样本利用mustang进行多结构比对,获得多结构比对信息;提取多结构比对信息中完全匹配的片段,形成该家族模板的折叠核心结构;对折叠核心片段进行骨架结构建模,形成家族模板;利用上述方法,对989种蛋白质折叠类型涵盖的3941家族分别构建家族模板,形成蛋白质家族模板数据库。
作为优选,所述折叠类型模板数据库构建为:蛋白质折叠类型模板以家族模板为单位通过系统聚类并经过筛选和验证最终得到;其中,
所述系统聚类方法为:对任意蛋白质折叠类型所属的n个家族模板,先将n个家族模板看成不同的n类,然后将性质最接近的两类合并为一类,再从n-1类中找到最接近的两类加以合并,依此类推,直到所有的家族模板被合为一类,得到n个家族模板的系统聚类图;家族模板通过tm-align进行两两比对,以tm-score作为距离参数,将tm-score取值最大的两家族合并;
所述任意蛋白质折叠类型i模板筛选的经验标准为:具有折叠类型i特有全部折叠核心片段,分布于系统聚类图中的独立分支,由家族模板首次合并形成,对蛋白质折叠类型i所属样本的识别率不低于80%;
利用上述方法,对989种蛋白质折叠类型分别构建模板,组成折叠类型模板数据库。
作为优选,tm-align可进行蛋白样本间的结构比对分析,所得打分值tm-score作为折叠类型模板构建的系统聚类参数,打分函数tm-score(templatemodelscore,模板建模打分)定义为:
其中,l是模板蛋白的长度,lali是模板蛋白与待测蛋白中匹配上的残基数目,di是模板蛋白与待测蛋白质中第i个匹配残基之间的距离,d0是作为标准化的距离参数,从而消除了打分值与蛋白质大小的幂率关系。
本发明的上述技术方案有如下优点:
1、家族模板的构建摒弃了从家族样本中选取天然结构样本作为模板,提高了家族模板的合理性以及适用性。
2、折叠类型模板构建以家族模板为单位通过系统聚类并经过筛选和验证最终得到,增加了折叠类型模板的可靠性。
3、分类方法取最大tm-score值作为评判参数,克服了以tm-score阈值0.5作为分类评判参数的不严谨性。
附图说明
图1为家族模板数据库分布图;
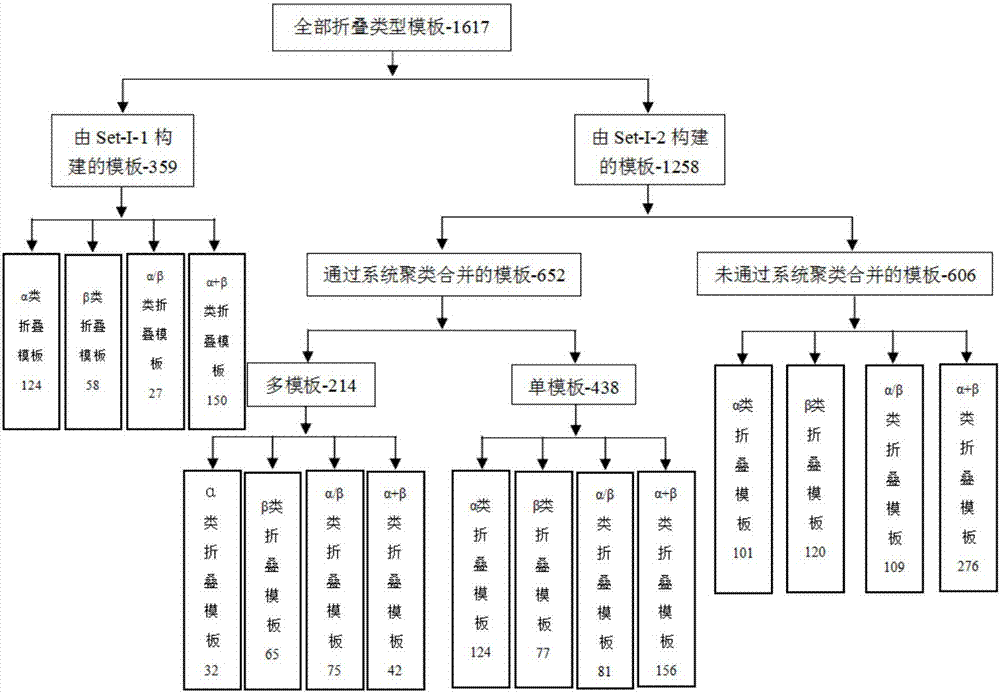
图2为折叠类型模板数据库分布图;
图3为蛋白质折叠类型的分类方法流程图。
具体实施方式
以下结合数据对本方法发明进行详细说明。
如图3所示,本发明实施例提供一种蛋白质折叠类型的分类方法包括以下步骤:
步骤1、构建模板数据库;
步骤2、基于模板数据库,将任意待测蛋白样本与模板数据中的所有模板进行tm-align比对,计算tm-score值,所述tm-score取值最大的模板所在的折叠类型即为待测蛋白样本所属折叠类型。具体过程包括如下:
一、材料的选取
本发明主要选取astralscope2.05数据库中相似性小于40%,且分辨率高于0.25nm的allalphaproteins(α),allbetaproteins(β),alphaandbetaproteins(α/β),alphaandbetaproteins(α+β)四类蛋白所属的折叠类型为研究对象,其中共有989种折叠类型、12165个样本,相应数据记为set-i。实验集中,有359种蛋白质折叠类型仅包含一个家族,且家族中仅包含一个样本,对于这部分折叠类型,需要利用astralscope2.05数据库中相似性小于95%的数据信息,相应数据记为set-i-1;其余630种蛋白质折叠类型含有两个及两个以上家族,对应的家族数及样本数分别为3582、11806,相应数据记为set-i-2。独立检验集:scopeastral2.06数据库中剔除scopeastral2.05所含样本,余下2142样本,涉及368种蛋白质折叠类型,记为set-ii。
二、家族模板设计方法及数据库的构建
家族模板设计方法的具体步骤为:对家族样本利用mustang进行多结构比对,获得多结构比对信息;提取多结构比对信息中完全匹配的片段(即家族样本共同参与的折叠核心片段),形成该家族模板的折叠核心结构;对折叠核心片段进行骨架结构建模(即提取骨架坐标信息),形成家族模板。
骨架坐标提取方法:对由n个样本组成的家族,利用mustang进行多结构比对,获得多结构比对结果,提取完全匹配片段,对匹配片段中任一残基i的α-碳原子匹配坐标信息--(xi,yi,zi),计算匹配坐标的平均值--
利用上述方法,对989种蛋白质折叠类型涵盖的3941家族分别构建家族模板,形成蛋白质家族模板数据库。数据库中的家族模板编号为scopeastral中相应家族代码,模板在四种结构类中的分布见图1。
三、折叠类型模板设计方法及数据库的构建
蛋白质折叠类型模板是以家族模板为单位通过系统聚类并经过筛选和验证最终得到。系统聚类的基本思想:对任意蛋白质折叠类型所属的n个家族模板,先将n个家族模板看成不同的n类,然后将性质最接近(距离最近)的两类合并为一类,再从n-1类中找到最接近的两类加以合并,依此类推,直到所有的家族模板被合为一类,得到n个家族模板的系统聚类图。家族模板通过tm-align进行两两比对,以tm-score作为距离参数,将tm-score取值最大(即距离最小)的两家族合并。
通过对系统聚类图中节点对应初始模板的计算分析及检验,提出任意蛋白质折叠类型i模板筛选的经验标准:具有折叠类型i特有全部折叠核心片段;分布于系统聚类图中的独立分支;由家族模板首次合并形成;对蛋白质折叠类型i所属样本的识别率不低于80%。
利用上述方法,对989种蛋白质折叠类型分别构建模板,组成折叠类型模板数据库,模板分布见图2。其中,由数据集set-i-1构建的模板359种,由于这些蛋白质折叠类型仅含一个家族,家族模板即为折叠类型模板;由数据集set-i-2构建的模板数共1258,其中508种蛋白质折叠类型成功筛选到了模板,另外的122种折叠类型未能筛选到满足条件的模板,以家族模板替代折叠类型模板。
四、分类方法的构建
将任意待测蛋白样本与模板数据中的所有模板进行tm-align比对,计算tm-score值。tm-score取值最大的模板所在的折叠类型即为待测蛋白样本所属折叠类型。
分类结果利用敏感性、特异性、matthew相关系数三个指标对其进行评估,参数定义如下:
敏感性:
特异性:
相关系数:
式中tp为真阳性个数,tn为真阴性个数,fp为假阳性个数,为fn假阴性个数。
五、分类效果
为验证模板设计及分类方法的合理性,以数据集set-i中的样本为研究对象,分别利用家族模板数据库与折叠类型模板数据库进行蛋白质折叠类型分类的自洽性检验,检验结果见表3与表4。s表示折叠类型所含样本数量,s'为真阳性与假阳性数量之和。
表3.家族模板的自洽性检验
表4.折叠类型模板的自洽性检验
由检验结果可知,基于家族模板数据库自洽性检验结果的敏感性、特异性及mcc的均值分别高达95.00%、99.99%、0.94,基于折叠类型模板数据库自洽性检验结果的敏感性、特异性以及mcc的均值分别为93.71%、99.97%及0.91。两种类型模板对相同数据集的分类检验结果相当,前者的分类结果略高后者。说明家族模板及折叠类型模板设计合理,模板反映了折叠类型的基本特征;前者的模板总数为3941,后者仅为1617,后者模板数仅为前者的五分之二,分类速度后者远远优于前者,分类精度家族模板略优于折叠类型模板。
为进一步检验模板数据库及分类方法的普适性,以数据集set-ii中的样本为研究对象,分别对家族模板数据库与折叠类型模板数据库进行独立性检验,检验结果见表5与表6。s+为数据集set-ii中样本数量。
表5.家族模板的独立性检验
表6.折叠类型模板的独立性检验
由上表可知,家族模板数据库及折叠类型模板数据库对扩充样本的分类效果稍差于自洽性检验中的结果,但是在独立性检验中家族模板与折叠类型模板的分类效果普遍高于90%,说明模板数据库及其分类方法可用于对扩充蛋白样本进行折叠类型的分类,从而验证了模板设计及分类方法具有有效的普适性。
目前蛋白质折叠类型的分类基本靠专家完成,而且不同库的分类结果并不相同,因此迫切需要建立一个基于统一原理的蛋白质折叠类型分类方法及分类模板数据库。本发明提供一种蛋白质折叠类型分类的方法,基于astralscope2.05数据库中相似性小于40%的α、β、α+β及α/β所属的折叠类型为研究对象,通过对蛋白质折叠结构分析及信息挖掘,建立了家族模板及蛋白质折叠类型模板设计方法,用于家族与折叠类型的模板设计,并完成了家族模板数据库与折叠类型模板数据库的构建,并建立基于模板的蛋白质折叠类型分类方法。使用本发明可实现蛋白质折叠类型的自动化分类。
- 还没有人留言评论。精彩留言会获得点赞!