一种基于图匹配的跨物种生物通路发现方法与流程
本发明属于图算法
技术领域:
,涉及一种基于图匹配的跨物种生物通路发现方法,尤其涉及一种跨物种生物蛋白质交互网络中生物通路的发现方法。
背景技术:
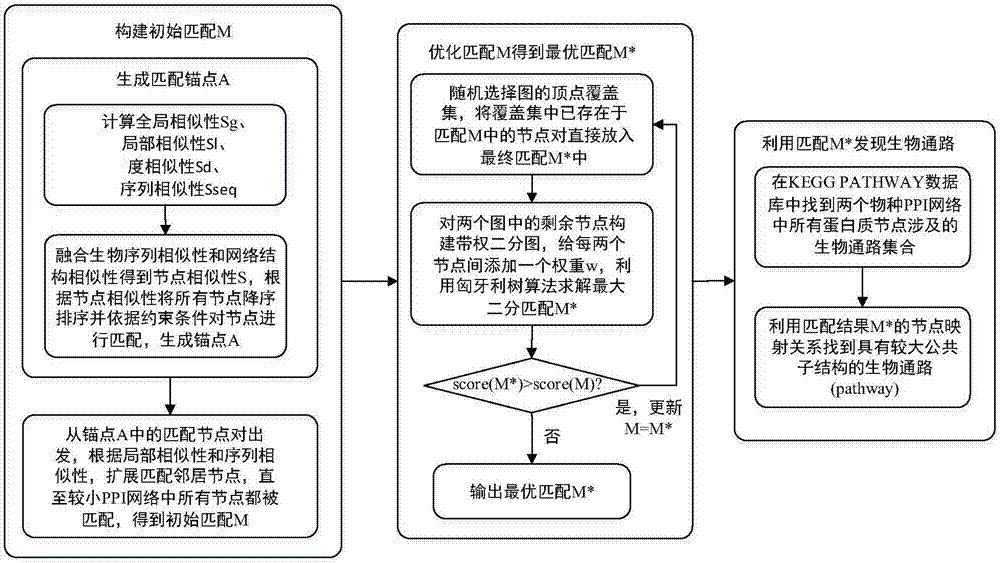
:图是计算机科学中常用的一类抽象数据结构,以描述事物之间的复杂关系。图结构已广泛应用于多种领域,如万维网、公路网、社交网络、知识图谱、蛋白质交互网络等。随着以上应用领域的发展,图数据不断的快速产生和积累,如何对其进行有效的管理、查询和挖掘等已成为学术界和工业界的研究热点。其中一个重要的课题就是挖掘不同物种生物蛋白质交互网络中的具有相似功能的生物通路。生物通路(biologicalpathway)由蛋白质和蛋白质之间的交互组成,可视为执行特定生物功能的最小的生物系统,不同物种之间存在大量相似功能的生物通路。有效挖掘发现不同物种之间具有相似功能的生物通路,可以帮助人类在生物体系层次上深刻理解物种间的相似及差别,对于基因学和医药学的发展具有重要的指导意义。传统生物通路发现方法需要大量的生物化学实验支撑,比较低效。通过图匹配方法,可以发现不同物种生物蛋白质交互网络(ppi网络)中相似结构和功能的子结构,根据这些匹配的子结构再根据生物化学方法验证它们是否是真正具有相似功能的生物通路,就比较有针对性和高效。图匹配问题目标是从两个由节点和边构成的图中得到节点一一映射的关系,实质上是图论中的子图同构问题,本身就是一个无法在多项式时间内解决的问题,随着生物蛋白质交互网络规模的扩大,蛋白质网络匹配问题面临着更加艰巨的挑战。早期蛋白质网络匹配技术主要采用序列匹配技术,因为蛋白质是由基因序列转码的mrna转译合成的,所以蛋白质也携带有基因序列信息,通过利用blast等序列匹配算法可以将序列信息相近的蛋白质进行匹配,然而只依赖蛋白质序列信息而忽略蛋白质网络的结构特性,导致匹配的准确性不高。后来pathblast、mawish、graemlin等启发式算法开始采用计算局部网络相似性来进行蛋白质网络匹配,但这些局部匹配算法可能会造成误导,因为一个物种的蛋白质网络子结构可能会匹配到另一个物种的蛋白质网络中的多个子结构,这种一对多的关系给确定生物通路带来了困难。现在比较流行的是采用全局网络匹配算法,比如isorank、path、ga、graal、l-graal、natalie、ghost、netal、magna、spinal、hubalign等。全局网络匹配算法强调两个蛋白质网络匹配的一对一映射关系,当且仅当两个蛋白质节点的邻居节点也匹配的情况下才会将两个蛋白质节点进行匹配。但现有全局网络匹配算法存在一个弊端,即进行网络匹配时不能很好地将蛋白质序列信息和蛋白质网络结构信息结合起来,造成序列信息匹配度和结构信息匹配度两者的对立,导致比较差的生物通路发现效果。技术实现要素:为了解决上述技术问题,本发明提供了一种基于新的图匹配算法的跨物种生物通路发现方法。本发明所采用的技术方案是:一种基于图匹配的跨物种生物通路发现方法,其特征在于,包括以下步骤:步骤1:构建初始匹配(matchingconstruction)阶段,通过结合各种相似性度量得到一个初始匹配方案,具体是:首先将两个物种的蛋白质交互网络(ppi网络)g1和g2中具有最高全局相似性、局部相似性、序列相似性和度相似性以及度数大于一定阈值的的节点进行匹配作为锚点a,然后从这些锚点a扩展,根据局部相似性和序列相似性匹配锚点的邻居节点,直至两个物种中节点总数较少的ppi网络中的所有节点均已匹配,得到初始匹配m。具体实现包括以下子步骤;步骤1.1:采用谱方法计算全局相似性sg;对于图g,它的邻接矩阵为a,对角度矩阵为d,拉普拉斯矩阵l=d-a;对于两个物种的蛋白质交互网络g1和g2,分别计算它们的拉普拉斯矩阵的特征值,假设图g1的拉普拉斯矩阵l1的特征值为α1≥α2≥…≥αn,图g2的拉普拉斯矩阵l2的特征值为β1≥β2≥…≥βn,令λ1=diag(αi),λ2=diag(βi),l1和l2是对称半正定矩阵,则其中u1和u2是正交矩阵;如果g1和g2是同构的,则存在一个列矩阵p使得pl1pt=l2,解得则全局相似性步骤1.2:计算局部相似性sl;假设图g中的顶点v的k步邻居子图为nk(v),并且表示包括节点v的完整k步子图,v1表示图g1的顶点集合,v2表示图g2的顶点集合,节点u∈v1和节点v∈v2两者之间的局部相似性通过比较u和v的k步邻居子图来衡量,具体如下:假设d(u)、d(v)分别为节点u和节点v在g1、g2中的度,假设nk(u)的所有节点的度按大小降序排列后分别为d1,1,d1,2,…,nk(v)的所有节点的度按大小降序排列后分别为d2,1,d2,2,…;令节点u和节点v的k步子图的较小节点总数nmin=min{|nk(u)|,|nk(v)|},则g1的节点u和g2的节点v之间的局部相似性为其中和分别表示包含节点u的k步子图的顶点数和边数,和分别表示包含节点v的k步子图的顶点数和边数,其中k步子图的最小度之和步骤1.3:计算度相似性sd和序列相似性sseq;假设g1的节点u的度为d(u),g2的节点v的度为d(v),那么u和v之间的度相似性为通过blast计算出序列分数seq(u,v),然后进行归一化得到序列相似性步骤1.4:融合各种相似性,选择出锚点a;首先由全局相似性矩阵sg、局部相似性矩阵sl得到拓扑相似性矩阵st=sg×sl,再结合度相似性sd得到网络结构相似性sstr(u,v)=(1-θ)×st(u,v)+θ×sd(u,v),其中θ表示拓扑相似性和度相似性的平衡参数,值在[0,1]之间可调节。再结合结构相似性sstr和序列相似性sseq得到最终的节点相似性s(u,v)=(1-α)×sstr(u,v)+α×sseq(u,v),其中α表示平衡结构相似性和度列相似性的平衡参数,值在[0,1]之间可调节。然后根据节点间的相似性分数s(u,v)将所有节点对进行降序排列,当节点对(u,v)满足以下两个条件时则将这两个节点进行匹配并加入锚点集合a;条件1:条件2:s(u,v)≥τ,其中τ是阈值;步骤1.5:从选择出的锚点集合a出发将邻居节点进行扩展匹配,得到初始匹配m;首先将锚点集合a中的匹配节点对都加入初始匹配m中,然后将锚点集合a中的所有匹配节点对(u,v)的邻居节点的笛卡尔积节点对(n(u)×n(v))加入优先队列q中,并按照扩展相似性se(u,v)=(1-α)×sl(u,v)+α×sseq(u,v)大小降序排列,然后逐个出列,如果扩展相似性最大的节点对(u,v)之前都没有与其他节点匹配过,那么将(u,v)匹配对加入m中,并将(u,v)的邻居节点笛卡尔积加入优先队列q中,直至队列q为空,得到初始匹配m。步骤2:优化匹配m得到最优匹配m*;匹配优化(matchingrefinement)阶段,步骤1中根据启发式算法得到的初始匹配m不能保证是最优的,采用一种新提出的算法进行匹配优化,具体是:首先从两个物种其中之一的蛋白质交互网络g1中随机选择一个顶点覆盖集c,然后保留顶点覆盖集c和初始匹配m的节点交集f1,以及保留f1在m中对应的g2的节点集合f2,对g1中不属于f1集合的节点和g2中不属于f2集合的节点进行匹配优化,得到优化后匹配m*,如果m*的匹配效果比m好,则更新m,如此多次迭代优化直至m不再更新,就得到接近最优的最终匹配结果。其中对g1中不属于f1集合的节点和g2中不属于f2集合的节点进行匹配优化,得到优化后匹配m*,具体实现包括以下子步骤:步骤2.1:构建一个带权二分匹配图gb,一边是包含v1-f1的节点结合,另一边是包含v2-f2的节点集合,对于所有u∈v1-f1和v∈v2-f2,在gb中增加一条边,边(u,v)的权重为w(u,v)=|m[n(u)∩f1]∩(n(v)∩f2)|;步骤2.2:利用匈牙利树算法计算出gb的最大带权二分匹配mb,使得二分图中所有边的权重之和最大化,那么优化后的匹配m*=(m∩(f1×f2))∪mb。判断匹配是否更好的标准是:其中如果u1和u2之间存在边,则为1,否则为0;如果v1和v2之间存在边,则为1,否则为0。步骤3:利用匹配m*发现生物通道;利用步骤2生成的两个物种的蛋白质交互网络匹配结果,结合生物数据库挖掘两个物种间共存的生物通路。keggpathway数据库中存储了现有实验证实的生物通路,其中一个生物通路(例如hsa03010)的表示是由物种代号(hsa表示人类)和一个数字组成(03010),相同数字的生物通路表示具有相似的生物功能。具体是:首先在keggpathway数据库中找到两个物种的蛋白质交互网络(ppi网络)中分别涉及到的所有生物通路,因为相同数字的生物通路表示具有相似的生物功能,所以可以得到两个物种中具有相似生物功能的生物通路集合,然后结合步骤2中生成的匹配结果,可以得到两个物种的具有相似生物功能的生物通路之间的结构映射关系,这个结构映射关系对于生命科学家研究不同物种之间生物通路之间的关联性具有指导意义。本发明具有以下优点:图匹配算法将生物序列相似性和网络结构相似性很好地融合,能够发现两个物种的蛋白质交互网络中共存的较大子结构,从而更有效地发现存在于不同物种中具有相似功能的生物通路。附图说明:图1是本发明实施例的流程图;图2是本发明实施例在样本图上匹配之后发现的共存的生物通路子结构,其中(1)hsa03010生物通路,(2)sce03010生物通路;图3是本发明实施例在样本图上发现的共存生物通路被apiddataserver证实的子结构,其中(1)hsa03010生物通路,(2)sce03010生物通路。具体实施方式为了便于本领域普通技术人员理解和实施本发明,下面结合附图及实施例对本发明作进一步的详细描述,应当理解,此处所描述的实施示例仅用于说明和解释本发明,并不用于限定本发明。本发明主要基于一种新提出的图匹配算法,将不同物种的蛋白质交互网络(ppi网络)进行最优匹配,根据匹配的结果在keggpathway数据库中进行查询,找到物种间共存的生物通路子结构。通过本发明,我们提供了一种新的跨物种生物通路发现方法,比传统生物化学方法更加高效,且算法效果比现有匹配算法更好。本发明提供的方法能够用计算机软件技术实现流程。参见图1,实施例以人类(human,生物代号hsapiens)的蛋白质交互网络(ppi网络)和酵母菌(yeast,生物代号scerevisiae)的蛋白质交互网络(ppi网络)为例,样本图的属性信息参见表1,表1样本图数据(人类和酵母菌的ppi网络)ppinetwork#nodes#edges#averagedegreehsapiens(human)1327611052816.651scerevisiae(yeast)58317714926.462本发明的具体实现包括以下步骤:步骤1:首先通过计算融合全局相似性、局部相似性、度相似性和序列相似性,得到人类和酵母菌的ppi网络中所有节点间的节点相似性,然后根据节点相似性将所有节点对降序排列,将满足要求的节点对加入锚点集合。然后从锚点集合中的节点对出发,根据局部相似性和序列相似性,扩展匹配它们的邻居节点,直至较小ppi网络中所有节点都被匹配,得到初始匹配。实施例中构建初始匹配的具体实施过程如下:首先,计算全局相似性sg、局部相似性sl、度相似性sd和序列相似性sseq,并融合为节点相似性s。计算全局相似性sg的方法为:先计算得到人类ppi网络g1和酵母菌ppi网络g2的拉普拉斯矩阵l1和l2,然后计算本征值,得到对角矩阵λ1和λ2,从而根据和得到u1和u2,则计算局部相似性sl的方法为:先计算得到g1中节点u和g2中节点v的k步邻居子图nk(u)和nk(v),然后根据
发明内容中提到的计算式计算得到。计算度相似性sd的方法为:先计算得到g1中节点u和g2中节点v的节点度d(u)和d(v),然后由计算得到。计算序列相似性sseq的方法为:将blast算法计算得到的序列相似性归一化即可。融合各个相似性为节点相似性s的方法为:s(u,v)=(1-α)×sstr(u,v)+α×sseq(u,v),其中sstr(u,v)=(1-θ)×st(u,v)+θ×sd(u,v),且st=sg×sl,α和β的值均为[0,1]之间可调节,这里均选取为0.5。然后,将所有节点对根据融合后的节点相似性s大小降序排列,将满足约束条件的节点对加入锚点集合a。这里约束条件为:(1)(2)s(u,v)≥τ,其中τ是满足不小于0.5的阈值,这里选取为0.5。最终,从锚点集合a中的每个节点对出发,扩展匹配它们的邻居节点,直至g1和g2中较小图中的所有节点均被匹配,即得到初始匹配m。扩展匹配的具体过程如下:将锚点集合a中的所有匹配节点对(u,v)的邻居节点的笛卡尔积节点对(n(u)×n(v))加入优先队列q中,并按照扩展相似性se(u,v)=(1-α)×sl(u,v)+α×sseq(u,v)大小降序排列,然后逐个出列,如果扩展相似性最大的节点对(u,v)之前都没有与其他节点匹配过,那么将(u,v)匹配对加入m中,并将(u,v)的邻居节点笛卡尔积加入优先队列q中,直至队列q为空,即得到初始匹配m。步骤2:上述启发式算法得到的初始匹配m不一定是最优的,继续将初始匹配m采用覆盖集和匈牙利树二分匹配算法进行优化得到最优匹配m*。实施例中优化初始匹配m得到最有匹配m*的具体实施过程如下:首先,随机选取g1的一个顶点覆盖集c,假设初始匹配m中所有g1节点集合为p1,令f1=c∩p1,f1中所有节点由初始匹配m映射的在g2中的节点结合为f2。保留f1和f2的匹配节点对直接加入到匹配m*中。然后,构建一个带权二分图gb,一边是所有v1-f1的节点集合,另一边是所有v2-f2的节点集合,二分图所有节点之间加一条边,边的权重为w(u,v)=|m[n(u)∩f1]∩(n(v)∩f2)|。然后利用匈牙利树算法(hungarianalgorithm)求解最大二分匹配问题,将计算的解,即所有剩余节点的匹配对加入到匹配m*中。最终,如果m*是比m更好的匹配,那么更新m=m*,然后重复优化匹配m的所有步骤,直至m*达到稳定。这里判断匹配是否更好的标准是:其中如果u1和u2之间存在边则为1,否则为0,同理。步骤3:在keggpathway数据库中查询人类ppi网络g1和酵母菌ppi网络g2各自涉及到的所有生物通路(pathway),然后根据最优匹配m*找到其中具有最大公共子结构的生物通路。图2给出了keggpathway中查询到的人类代号为hsa03010的生物通路,共包含132个蛋白质节点和1924条边,还有查询到的酵母菌代号为sce03010的生物通路,共包含175个蛋白质节点和2311条边,其中最优匹配m*发现了hsa03010和sce03010的最大公共子结构包含63个蛋白质分子和1406条边,该公共子结构在图2中分别均以深色标出。hsa03010和sce03010具有相同的数字标号03010,表明人类和酵母菌的这两个生物通路具有相似的生物功能。为了进一步证明人类生物通路hsa03010和酵母菌生物通路sce03010之间的关联性,我们采用apiddataserver查询这两个生物通路中已被生物实验证实确实存在的蛋白质及蛋白质之间的联系。apid查询结果表明hsa03010生物通路中总共有126个蛋白质节点和1748条边是被实验证实确实存在的,sce03010生物通路中总共有165个蛋白质节点和192条边是被实验证实确实存在的,它们之间存在一个由26个蛋白质节点和32条边构成的较为完整的公共子结构,该结构展示在图3中。该结果说明本发明提出的图匹配方法可以有效地发现人类和酵母菌中具有相似功能的生物通路公共子结构,该结果对于生物学研究物种之间的联系有指导意义。应当理解的是,本说明书未详细阐述的部分均属于现有技术。应当理解的是,上述针对较佳实施例的描述较为详细,并不能因此而认为是对本发明专利保护范围的限制,本领域的普通技术人员在本发明的启示下,在不脱离本发明权利要求所保护的范围情况下,还可以做出替换或变形,均落入本发明的保护范围之内,本发明的请求保护范围应以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1