基于二代测序的肿瘤克隆变异检测方法、装置和存储介质与流程
本申请涉及肿瘤检测领域,特别是涉及一种基于二代测序的肿瘤克隆变异检测方法、装置和存储介质。
背景技术
肿瘤是由基因组变异引起的疾病。肿瘤的发生伴随着大量的基因组变异,这些基因组变异根据发生的时期不同分为两种肿瘤克隆变异类型,即克隆性突变和亚克隆性突变。克隆性的突变,发生在肿瘤早期,存在于大部分的肿瘤细胞中;亚克隆性的突变,发生在肿瘤后来的进化过程中,存在于小部分的肿瘤细胞亚群中。这些克隆性突变和亚克隆性突变组成了肿瘤内部的异质性,与肿瘤的进化,转移,复发,耐药相关。
目前肿瘤克隆性研究可以使用的技术有:二代测序技术、snp芯片、微阵列比较基因组杂交技术等。单细胞测序技术可以直接观察到单个细胞水平的异质性,但是单细胞技术仍然面临很大的挑战,比如等位基因脱扣(alleledropout,ado)、扩增失败和偏性扩增等现象的存在,导致单细胞测序研究肿瘤异质性不成熟。因此,利用混在一起的细胞群仍然是研究肿瘤克隆性变异的有效方法。大部分的实体瘤和血液肿瘤都存在不同程度的异质性,基本都是由一个主克隆和几个亚克隆组成。
很多研究都显示肿瘤内部的异质性和克隆组成具有临床意义,并且促进了肿瘤治疗的耐药发生。一些研究发现肿瘤中的亚克隆细胞的出现与慢性淋巴细胞白血病的不良预后相关,与食管癌和黑色素瘤的恶性进展相关。亚克隆突变还与耐药相关,例如在非小细胞肺癌中经常发生egfr的亚克隆突变。针对亚克隆突变也有靶向药物的开发,例如奥西替尼就是针对egfrt790m的亚克隆突变。因此,要开发二线治疗的靶向药物就必须了解肿瘤的克隆进化过程,了解肿瘤克隆变异类型,以及与肿瘤治疗的分子机理。
目前研究肿瘤克隆性一般是取一个患者身上多个时间点或多处肿瘤组织进行研究,一次的样本数在6~20个左右,这样就限制了克隆研究的样本。因此利用二代测序技术和生物信息学方法,能够推测一个时间节点的样本,这样就使克隆研究的适用范围变广。针对单个时间节点的肿瘤样本进行克隆性研究,一般采取的方法是根据变异在测序数据中的信号的频率来判断是属于克隆还是亚克隆,即根据变异信号的频率来判断肿瘤克隆变异类型。但是,这些方法并不能很准确的检测出肿瘤克隆变异类型,很难直接用于二线治疗的靶向药物开发或临床用药指导。
技术实现要素:
本申请的目的是提供一种新的基于二代测序的肿瘤克隆变异检测方法、装置和存储介质。
为了实现上述目的,本申请采用了以下技术方案:
本申请的第一方面公开了一种基于二代测序的肿瘤克隆变异检测方法,包括以下步骤,
肿瘤突变频率鉴定步骤,包括对成对的肿瘤和正常样本的测序结果的比对文件进行突变检测,获取突变的测序片段支持数、正常的测序片段支持数和总的测序片段支持数;并计算肿瘤突变频率,即突变的测序片段支持数除以总的测序片段支持数,获得肿瘤突变频率;
肿瘤样本纯度鉴定步骤,包括获取肿瘤和正常样本中的每个snp位点两种碱基的测序片段支持数,将碱基频率小于或大于设定阈值的snp位点定义为纯合位点,将剔除纯合位点的snp的信息,转化为纯度检测软件的输入数据集,得到肿瘤样本纯度鉴定结果和拷贝数信息;
肿瘤拷贝数鉴定步骤,包括对经过纯度矫正的拷贝数信息及相应区域进行过滤筛选,并将小片段合并成大片段,对突变区域的拷贝数进行注释,获得肿瘤拷贝数鉴定结果;
肿瘤突变频率校正步骤,包括根据肿瘤样本纯度鉴定步骤和肿瘤拷贝数鉴定步骤的结果,利用beta分布模型计算突变细胞在所测肿瘤组织中的比例,获得校正后的肿瘤突变频率;
肿瘤克隆变异类型鉴定步骤,包括根据所述校正后的肿瘤突变频率判断突变类型的克隆属性,获得肿瘤克隆变异结果。
需要说明的是,本申请针对现有的肿瘤克隆变异类型检测不够准确的问题,创造性的在肿瘤克隆变异类型的检测过程中,引入肿瘤样本纯度鉴定和肿瘤拷贝数鉴定,通过突变的样本纯度和拷贝数对突变频率进行校正,从而可以获得更准确的突变细胞比例计算结果,进而获得更准确的肿瘤克隆变异类型检测结果;本申请的检测方法避免了样本纯度问题和倍型问题对肿瘤克隆变异类型检测结果的影响,使得肿瘤克隆变异类型检测结果更加准确,从而可以直接用于二线治疗的靶向药物开发或临床用药指导。
优选的,肿瘤突变频率鉴定步骤中,突变检测包括点突变、短片段的插入缺失和/或杂合性缺失。并且优先选取测序质量高的片段作为最终的统计结果。
优选的,肿瘤突变频率鉴定步骤中,还包括矫正的步骤,所述矫正的步骤包括,若两条成对序列在重叠区域里碱基类型一致,则只保留区域里质量值较高的一条序列;若碱基类型不一致,并且其中一条序列质量高,另一条质量低,则保留质量高的序列,否则两条都舍弃。
需要说明的是,因为二代测序的片段都是成对的,一对片段之间的插入片段区域如果较小,则两条片段都会覆盖到突变位置,这样的两条片段实际上为一条片段,因此需要矫正,需要对成对序列重叠区域的进行处理;所以,本申请的优选方案中进一步引入了矫正的步骤。
优选的,肿瘤样本纯度鉴定步骤中,将碱基频率小于30%或大于70%的snp位点定义为纯合位点,去掉纯合位点后,将剩下的snp位点两种碱基的测序片段支持数通过公式转化为纯度检测软件输入需要的两种信号,进行纯度鉴定,具体采用公式(2-1)和公式(2-2);
其中,i表示snp位点,na,i表示i位点的a碱基的深度,nb,i表i位点的b碱基的深度,d表示肿瘤的平均深度,baf表示b碱基的频率。
优选的,肿瘤突变频率校正步骤中,计算突变细胞在所测肿瘤组织中的比例,具体采用公式(4-1)或公式(4-2),
其中,pdf(ccf,m)表示突变肿瘤细胞比例的密度分布函数,βpdf表示beta密度分布函数,ccf表示突变细胞在所测肿瘤组织中的比例,α为纯度,q(m)表示肿瘤的拷贝数,alt(m)是突变的测序片段支持数,ref(m)是正常的测序片段支持数。
需要说明的是,一般的肿瘤克隆变异采用公式(4-1)即可,如果需要考虑杂合型缺失(loh)的情况,则采用公式(4-2)。
优选的,肿瘤克隆变异类型鉴定步骤中,根据校正后的肿瘤突变频率判断突变类型的克隆属性,具体包括,突变细胞在所测肿瘤组织中的比例等于1的概率大于0.5,判断突变类型是克隆性突变,反之则为亚克隆性突变。
本申请的第二方面公开了一种基于二代测序的肿瘤克隆变异检测装置,包括,
肿瘤突变频率鉴定模块,用于对成对的肿瘤和正常样本的测序结果的比对文件进行突变检测,获取突变的测序片段支持数、正常的测序片段支持数和总的测序片段支持数;并计算肿瘤突变频率,即突变的测序片段支持数除以总的测序片段支持数,获得肿瘤突变频率;
肿瘤样本纯度鉴定模块,用于获取肿瘤和正常样本中的每个snp位点两种碱基的测序片段支持数,将碱基频率小于或大于设定阈值的snp位点定义为纯合位点,将剔除纯合位点的snp的信息,转化为纯度检测软件的输入数据集,得到肿瘤样本纯度鉴定结果和拷贝数信息;
肿瘤拷贝数鉴定模块,用于对经过纯度矫正的拷贝数信息及相应区域进行过滤筛选,并将小片段合并成大片段,对突变区域的拷贝数进行注释,获得肿瘤拷贝数鉴定结果;
肿瘤突变频率校正模块,用于根据肿瘤样本纯度鉴定步骤和肿瘤拷贝数鉴定步骤的结果,利用beta分布模型计算突变细胞在所测肿瘤组织中的比例,获得校正后的肿瘤突变频率;
肿瘤克隆变异类型鉴定模块,用于根据校正后的肿瘤突变频率判断突变类型的克隆属性,获得肿瘤克隆变异结果。
优选的,肿瘤样本纯度鉴定模块包括snparray上的纯度检测软件ascat。
本申请的第三方面公开了一种基于二代测序的肿瘤克隆变异检测装置,包括:
存储器,用于存储程序;
处理器,用于通过执行存储器存储的程序以实现本申请的肿瘤克隆变异检测方法。
本申请的第四方面公开了一种计算机可读存储介质,包括程序,该程序能够被处理器执行以实现本申请的肿瘤克隆变异检测方法。
由于采用以上技术方案,本申请的有益效果在于:
本申请的肿瘤克隆变异类型检测方法,避免了样本纯度问题和倍型问题对检测结果的影响,使得肿瘤克隆变异类型检测结果更加准确,能够识别有临床意义的亚克隆突变,为临床治疗和靶向药物研发提供了准确的科学依据;同时,也为进一步准确、深入的研究肿瘤克隆进化过程,研究肿瘤治疗分子机理奠定了基础。
附图说明
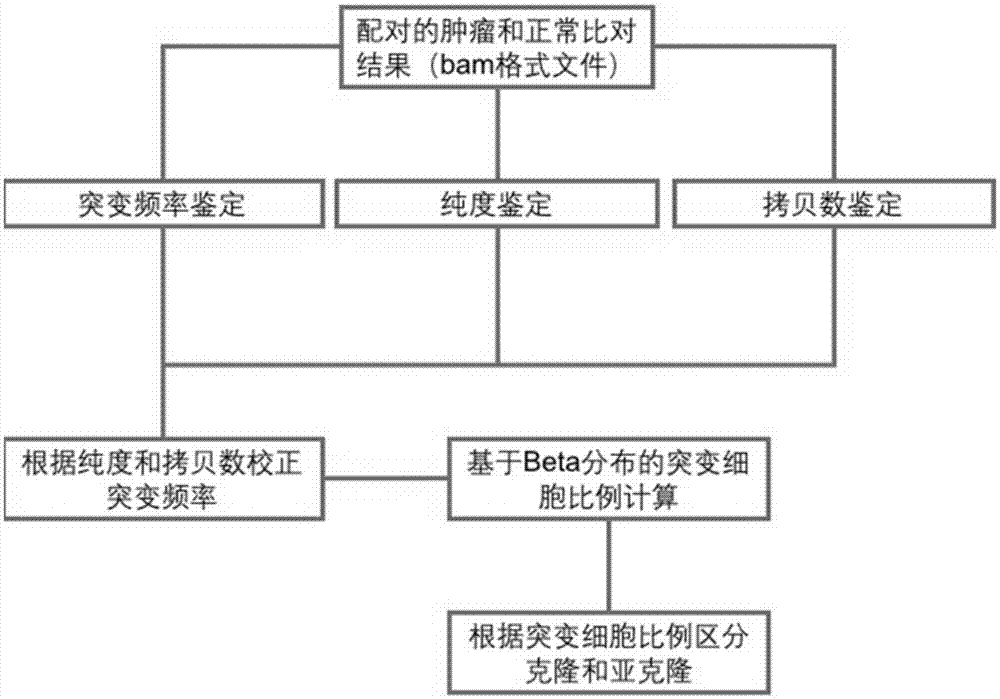
图1是本申请实施例中基于二代测序的肿瘤克隆变异检测方法的流程框图;
图2是本申请实施例中基于二代测序的肿瘤克隆变异检测装置的结构框图;
图3是本申请实施例中采用现有技术对一个非小细胞肺癌的外显子基因组的基于矫正前的突变频率进行肿瘤克隆变异类型检测的结果;
图4是本申请实施例中采用本申请的肿瘤克隆变异类型检测方法对一个非小细胞肺癌的外显子基因组的基于矫正后的突变频率进行肿瘤克隆变异类型检测的结果;
图5是本申请实施例中对肺癌患者进行肿瘤克隆变异类型检测的结果;
图6是本申请实施例中根据肺癌患者的肿瘤克隆变异类型检测结果进行免疫治疗的效果分析图。
具体实施方式
下面通过具体实施方式结合附图对本申请作进一步详细说明。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
本申请在对肿瘤克隆变异类型检测的研究过程中发现,现有的检测方法都是假定肿瘤倍型是二倍体,但是,实际上肿瘤有很大一部分是多倍体,不同区域会发生不同的拷贝数变异,甚至发生基因组加倍的现象,这使得现有的肿瘤克隆变异类型检测结果不能够准确的反映真实的变异克隆性情况。并且,在实际检测中,进行检测的肿瘤样本的纯度很少是100%,几乎都有不同程度的正常细胞污染,这也使得变异的克隆性分析不准确。
基于以上研究和认识,本申请创造性的提出,分别对二代测序结果进行突变及其频率鉴定、肿瘤样本纯度鉴定和肿瘤拷贝数鉴定,通过突变的样本纯度和拷贝数对突变频率进行校正,然后再基于beta分布模型计算突变细胞比例,进而判断肿瘤克隆变异类型。这样就很好的减小了样本纯度问题以及多倍体或基因组加倍等拷贝数变异对肿瘤克隆变异类型检测结果的影响,使得肿瘤克隆变异类型检测结果更加准确。
如图1所示,本申请的基于二代测序的肿瘤克隆变异检测方法,包括以下步骤,
(1)肿瘤突变频率鉴定步骤,包括对成对的肿瘤和正常样本的测序结果的比对文件进行突变检测,获取突变的测序片段支持数、正常的测序片段支持数和总的测序片段支持数;并计算肿瘤突变频率,即突变的测序片段支持数除以总的测序片段支持数,获得肿瘤突变频率。其中,变异检测软件包括但不仅限于常规使用的mutect、varscan等变异检测软件。突变检测包括点突变、短片段的插入缺失和/或杂合性缺失,选取测序质量高的片段作为最终的统计结果。
在一些实施例中,因为二代测序的片段都是成对的,一对片段之间的插入片段区域如果较小,则两条片段都会覆盖到突变位置,这样的两条片段实际上为一条片段,因此需要矫正。因此,突变频率鉴定步骤中还包括矫正的步骤,矫正的步骤包括,若两条成对序列在重叠区域里碱基类型一致,则只保留区域里质量值较高的一条序列;若碱基类型不一致,并且其中一条序列质量高,另一条质量低,则保留质量高的序列,否则两条都舍弃。
(2)肿瘤样本纯度鉴定步骤,包括获取肿瘤和正常样本中的每个snp位点两种碱基的测序片段支持数,将碱基频率小于或大于设定阈值的snp位点定义为纯合位点,将剔除纯合位点的snp的信息,转化为纯度检测软件的输入数据集,得到肿瘤样本纯度鉴定结果和拷贝数信息。
临床上估计肿瘤组织纯度的方法是取一点肿瘤组织放在显微镜下观察组织中明显是肿瘤细胞的比例。但是,由于测序样品与显微镜观察样品不是同一批样品,所以临床观察的纯度往往对信息分析的参考意义不大。
信息学上对纯度进行预测主要决定于两个因素,一个是碱基频率,另一个是拷贝数。有很大一部分癌症的基因组非常不稳定,会发生大量的拷贝数变异,甚至发生基因组加倍的现象,并且有很多研究找到了拷贝数变异与表达的关系,证实与癌症相关,所以估计纯度时的拷贝数因素不能忽略。
假设突变碱基的拷贝数为q,样品纯度是p,正常基因组的拷贝数为2,则碱基频率f具体采用公式(2)计算,
f=p*q/(p*(q+1)+2*(1-p))(2)
公式(2)中,
f为碱基频率,q为突变碱基的拷贝数,p为肿瘤样本的纯度。
本例具体的,利用snparray上的软件ascat,对成对样本进行纯度检测。ascat是应用在snparray上的软件,能从snp位点信息出发,预测拷贝数变异和纯度。由于二代测序技术的纯度预测工具还不成熟,很少有软件能够很好的预测肿瘤纯度。多项数据支持snparray与二代测序技术找出来的拷贝数变异结果一致率较高,因此参考ascat的预测方法,将二代测的信号转成snparray的信号。
在一些实施例中,选择突变位点tumor和正常位点normal中germline突变位点,将碱基频率小于30%或大于70%的snp位点定义为纯合位点,将踢掉纯合位点后的snp作为数据集,处理成snparray的类似形式。snparray中,用logr来表示位点总的信号强度,用baf(即ballelefrequency)表示b碱基的频率,假设snp位点有a、b两种碱基组成,则它们的关系表示如下面的公式(2-1)、公式(2-2)两个公式:
其中,i表示snp位点,na,i表示i位点的a碱基的深度,nb,i表i位点的b碱基的深度,d表示突变肿瘤的平均深度,baf表示b碱基的频率。将上面两个转化的信号作为ascat的输入,进行纯度和拷贝数的检测。
(3)肿瘤拷贝数鉴定步骤,包括对经过纯度矫正的拷贝数信息及相应区域进行过滤筛选,并将小片段合并成大片段,对突变区域的拷贝数进行注释,获得肿瘤拷贝数鉴定结果。
在一些实施例中,采用ascat的结果对拷贝数进行片段化,并且给出区域片段化的绝对拷贝数,这些拷贝数信息已经经过纯度的矫正。对这些区域进行过滤筛选,对小片段进行合并成大片段,对突变区域的拷贝数进行注释。根据突变的样本纯度和拷贝数结果校正“(1)肿瘤突变频率鉴定步骤”获得的肿瘤突变频率。
(4)肿瘤突变频率校正步骤,包括根据肿瘤样本纯度鉴定步骤和肿瘤拷贝数鉴定步骤的结果,利用beta分布模型计算突变细胞在所测肿瘤组织中的比例,获得校正后的肿瘤突变频率。
根据肿瘤纯度以及拷贝数,就可以精确的量化突变在所测肿瘤组织中的比例(cancercellfraction,缩写ccf),判断突变发生是属于克隆性clonal还是亚克隆subclonal。本例采用的是beta分布模型。
在一些实施例中,计算突变细胞在所测肿瘤组织中的比例,具体采用公式(4-1),
ccf的值从0~1,pdf(ccf,m)表示突变肿瘤细胞比例的密度分布函数,βpdf表示beta密度分布函数,ccf表示突变细胞在所测肿瘤组织中的比例,α为纯度,q(m)表示肿瘤的拷贝数,alt(m)是突变的测序片段支持数,ref(m)是正常的测序片段支持数。于是可以得到概率最高的ccf值。
在一些实施例中,还考虑了杂合型缺失(loh)的情况,在loh情况下,ccf的计算可以用公式(4-2)。
取概率高的ccf作为结果。
(5)肿瘤克隆变异类型鉴定步骤,包括根据校正后的肿瘤突变频率判断突变类型的克隆属性,获得肿瘤克隆变异结果。
在一些实施例中,认为ccf>0.8的概率如果大于0.5就认为是clonal,反之,是subclonal;另外在一些更为严谨的判断中,认为ccf=1的概率大于0.5,判断是clonal,反之,是subclonal。
本领域技术人员可以理解,上述实施方式方法的全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现上述功能。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现上述全部或部分功能。另外,当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器中的程序时,即可实现上述实施方式中全部或部分功能。
因此,如图2所示,本申请一实施例中,基于二代测序的肿瘤克隆变异检测装置,包括:肿瘤突变频率鉴定模块201,肿瘤样本纯度鉴定模块202,肿瘤拷贝数鉴定模块203,肿瘤突变频率校正模块204,和肿瘤克隆变异类型鉴定模块205。
其中,肿瘤突变频率鉴定模块201,用于对成对的肿瘤和正常样本的测序结果的比对文件进行突变检测,获取突变的测序片段支持数、正常的测序片段支持数和总的测序片段支持数;并计算肿瘤突变频率,即突变的测序片段支持数除以总的测序片段支持数,获得肿瘤突变频率;肿瘤样本纯度鉴定模块202,用于获取肿瘤和正常样本中的每个snp位点两种碱基的测序片段支持数,将碱基频率小于或大于设定阈值的snp位点定义为纯合位点,将剔除纯合位点的snp的信息,转化为纯度检测软件的输入数据集,得到肿瘤样本纯度鉴定结果和拷贝数信息;肿瘤拷贝数鉴定模块203,用于对经过纯度矫正的拷贝数信息及相应区域进行过滤筛选,并将小片段合并成大片段,对突变区域的拷贝数进行注释,获得肿瘤拷贝数鉴定结果;肿瘤突变频率校正模块204,用于根据肿瘤样本纯度鉴定步骤和肿瘤拷贝数鉴定步骤的结果,利用beta分布模型计算突变细胞在所测肿瘤组织中的比例,获得校正后的肿瘤突变频率;肿瘤克隆变异类型鉴定模块205,用于根据校正后的肿瘤突变频率判断突变类型的克隆属性,获得肿瘤克隆变异结果。
本申请另一实施例还提供一种基于二代测序的肿瘤克隆变异检测装置,包括:存储器,用于存储程序;处理器,用于通过执行上述存储器存储的程序以实现如下方法:肿瘤突变频率鉴定步骤,包括对成对的肿瘤和正常样本的测序结果的比对文件进行突变检测,获取突变的测序片段支持数、正常的测序片段支持数和总的测序片段支持数;并计算肿瘤突变频率,即突变的测序片段支持数除以总的测序片段支持数,获得肿瘤突变频率;肿瘤样本纯度鉴定步骤,包括获取肿瘤和正常样本中的每个snp位点两种碱基的测序片段支持数,将碱基频率小于或大于设定阈值的snp位点定义为纯合位点,将剔除纯合位点的snp的信息,转化为纯度检测软件的输入数据集,得到肿瘤样本纯度鉴定结果和拷贝数信息;肿瘤拷贝数鉴定步骤,包括对经过纯度矫正的拷贝数信息及相应区域进行过滤筛选,并将小片段合并成大片段,对突变区域的拷贝数进行注释,获得肿瘤拷贝数鉴定结果;肿瘤突变频率校正步骤,包括根据肿瘤样本纯度鉴定步骤和肿瘤拷贝数鉴定步骤的结果,利用beta分布模型计算突变细胞在所测肿瘤组织中的比例,获得校正后的肿瘤突变频率;肿瘤克隆变异类型鉴定步骤,包括根据校正后的肿瘤突变频率判断突变类型的克隆属性,获得肿瘤克隆变异结果。
本申请另一种实施例还提供一种计算机可读存储介质,包括程序,该程序能够被处理器执行以实现如下方法:肿瘤突变频率鉴定步骤,包括对成对的肿瘤和正常样本的测序结果的比对文件进行突变检测,获取突变的测序片段支持数、正常的测序片段支持数和总的测序片段支持数;并计算肿瘤突变频率,即突变的测序片段支持数除以总的测序片段支持数,获得肿瘤突变频率;肿瘤样本纯度鉴定步骤,包括获取肿瘤和正常样本中的每个snp位点两种碱基的测序片段支持数,将碱基频率小于或大于设定阈值的snp位点定义为纯合位点,将剔除纯合位点的snp的信息,转化为纯度检测软件的输入数据集,得到肿瘤样本纯度鉴定结果和拷贝数信息;肿瘤拷贝数鉴定步骤,包括对经过纯度矫正的拷贝数信息及相应区域进行过滤筛选,并将小片段合并成大片段,对突变区域的拷贝数进行注释,获得肿瘤拷贝数鉴定结果;肿瘤突变频率校正步骤,包括根据肿瘤样本纯度鉴定步骤和肿瘤拷贝数鉴定步骤的结果,利用beta分布模型计算突变细胞在所测肿瘤组织中的比例,获得校正后的肿瘤突变频率;肿瘤克隆变异类型鉴定步骤,包括根据校正后的肿瘤突变频率判断突变类型的克隆属性,获得肿瘤克隆变异结果。
下面通过具体实施例和附图对本申请作进一步详细说明。以下实施例仅对本申请进行进一步说明,不应理解为对本申请的限制。
实施例
采用基于二代测序的肿瘤克隆变异检测方法,本例对一例非小细胞肺癌,获取了肿瘤组织和正常对照的外显子数据,外显子数据由深圳裕策生物科技有限公司提供,进行变异检测,拷贝数分析和纯度分析纠正后,基于突变频率信息进行克隆鉴定,结果如图4所示。与此同时,对相同的外显子数据采用现有技术,即不进行拷贝数分析和纯度分析纠正,直接由二代测序结果获得的突变频率分析克隆性,作为对照,结果如图3所示。
图3是突变频率经过拷贝数和纯度矫正前的克隆区分,即直接由二代测序结果直接用软件mutect获得的突变频率分析得到的克隆性分析结果图,图片展示软件为r(version3.3.2)。图4是参考本申请方法经过拷贝数和纯度矫正后的克隆区分,即按照本申请的方法由样本纯度和拷贝数结果校正突变频率后,根据基于beta分布模型得到的肿瘤克隆变异类型检测图,图片展示软件为r(version3.3.2)。比较图3和图4的结果显示,图3中的突变频率分布无法有效识别克隆类别,而图4中矫正后的突变频率能够识别克隆类别。
另外,采用基于二代测序的肿瘤克隆变异检测方法,本例进一步对一例肺癌患者的肿瘤样本进行肿瘤克隆变异类型检测,全外显子二代测序结果由深圳裕策生物科技有限公司提供。检测结果如图5所示,采用本例的基于二代测序的肿瘤克隆变异检测方法首先鉴定出了酪氨酸激酶抑制剂敏感性靶点egfrl858r的基因突变,同时鉴定出耐药性亚克隆突变egfrt790m。而对于egfrt790m突变的患者,需要用第三代抑制剂奥西替尼进行治疗。可见,本例的方法能够识别有临床意义的亚克隆突变。
基于以上检测结果,针对13例进行过免疫治疗的非小细胞肺癌患者,利用本例的基于二代测序的肿瘤克隆变异检测方法进行克隆突变鉴定,检测结果如图6所示;其中,13例非小细胞肺癌患者的外显子二代测序数据由深圳裕策生物科技有限公司提供。图6的结果显示,应用免疫治疗的肿瘤患者,疗效好比疗效差有更多的克隆性突变。可见,本例的肿瘤克隆变异类型检测方法,能够用于临床用药指导,提供肿瘤治疗效果。
以上内容是结合具体的实施方式对本申请所作的进一步详细说明,不能认定本申请的具体实施只局限于这些说明。对于本申请所属技术领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!