一种纳米单晶三元材料及其制备方法与流程
1.本发明涉及锂离子电池正极材料及其制备方法,属于锂离子电池正极材料领域,具体涉及一种纳米单晶三元材料及其制备方法。
背景技术:
2.锂离子电池目前广泛应用于新能源汽车领域。目前纯电动汽车和插电混动汽车较为成熟,而轻混合动力汽车由于对电池的放电倍率性能和浅充放电条件下超长循环性能的要求,目前一般采用镍氢电池方案,采用锂离子电池解决方案很难实现和车辆的寿命同步,目前的方案是车辆中期更换或者从设计妥协,增大电池组的容量来降低倍率解决相关问题。
3.对于纯电动汽车和插电混合动力汽车,动力电池的充放电倍率一般不超过10c。对于轻混合动力汽车,电池的工作工况与纯电动汽车和插电混动汽车有所不同。作为唯一的能量来源,纯电动汽车的电池系统一般在较大荷电状态范围内进行充放电,以保证续航里程,这种使用工况下,由于电池组能量较大,电池的使用放电倍率一般不超过5c,充电倍率一般不超过3c,普通的微米级小颗粒单晶材料和二次颗粒材料可以满足使用要求。对于插电混合动力汽车,使用工况分为短途的纯电动通行和长途的混合动力通行,对于短途纯电通行,电池的使用工况与纯电动汽车基本一致,一般放电倍率不超过10c,充电倍率不超过5c,对于长途混合动力通行,使用工况接近于轻混合动力汽车,即电池保持在一定荷电状态,此时的充放电为一般不超过30s的瞬间充放电,倍率也一般不超过10c。对于轻混合动力汽车,电池主要工作在平衡荷电状态下,需要以较大的倍率进行瞬间充放电,主要是在车辆加速时提供能量,在减速时回收能量,在这种工况下,电池需要有非常好的倍率性能和循环性能,如果与车辆同寿命,这种在相对恒定的荷电状态下的浅充放循环需要百万次数量级,充放电倍率需要100c数量级。在这种使用条件下,目前的绝大部分二次颗粒三元材料和微米级单晶三元材料不能满足使用要求。微米级的单晶材料在倍率上首先无法达到使用要求,微米级的硬团聚二次颗粒材料,在100c充放电的情况下进行长期浅充放电循环过程中,材料应力集中,在一次颗粒之间形成断裂,生成新的表面,新的表面在大倍率充放电的过程中与电解液发生副反应,消耗电解液同时加速电池正极一侧内阻的增长,使电池的性能跳水,混合动力系统失效,这个过程很难达到100万次数量级。
技术实现要素:
4.基于上述技术背景,本发明人进行了锐意进取,结果发现:通过将含镍钴锰的前驱体、锂源、含a元素的化合物和含m元素的化合物经研磨混合后烧结、粉碎、湿法包覆、喷雾干燥,最后经过烧结制得本发明所述的纳米单晶三元材料,本发明采用喷雾的方法生成松散的二次团聚体,由于其为软团聚,可以在电池浆料匀浆过程中被打开,在循环过程中不存在应力集中的问题,减少开裂可能性,同时通过湿法均匀的包覆,纳米颗粒表面形成包覆层,可实现锂离子快速传导,从而提高材料的倍率性能和循环性能,本发明所述纳米单晶三元
材料的制备方法简单,在制备过程中对湿度控制要求降低,成本较低。
5.本发明的第一方面在于提供一种纳米单晶三元材料,所述纳米单晶三元材料由前驱体、锂源和含a元素的化合物混合烧结后与含m元素的化合物湿法包覆,最后经喷雾干燥制得;
6.所述a元素选自mg、al、ti和zr中的一种或几种;
7.所述m元素选自si、al和ti中的一种或几种。
8.本发明的第二方面在于提供一种根据本发明第一方面所述的纳米单晶三元材料的制备方法,该制备方法包括以下步骤:
9.步骤1、将称量好的锂源、预处理后的前驱体、含a元素的化合物湿法研磨、混合、干燥;
10.步骤2、将步骤1制得混合物高温烧结后粉碎;
11.步骤3、步骤2烧结后的材料与含m元素的化合物进行湿法包覆后喷雾干燥;
12.步骤4、将步骤3制得材料烧结。
13.本发明提供的纳米单晶三元材料及其制备方法具有以下优势:
14.(1)本发明所述的纳米单晶三元材料具备较好的物理储存性能,对材料生产制造和材料制成电池过程中,对工艺特别是湿度控制要求降低;
15.(2)本发明所述的纳米单晶三元材料具有更好的倍率性能和循环性能;
16.(3)本发明所述的纳米单晶三元材料的制备方法简单,制备成本较低。
附图说明
17.图1-a示出本发明实施例1制得产物的扫描电镜照片;
18.图1-b示出本发明实施例1制得产物的扫描电镜放大照片;
19.图2-a示出本发明实施例2制得产物的扫描电镜照片;
20.图2-b示出本发明实施例2制得产物的扫描电镜放大照片;
21.图3-a示出本发明实施例3制得产物的扫描电镜照片;
22.图3-b示出本发明实施例3制得产物的扫描电镜放大照片;
23.图4-a示出本发明对比例1制得产物的扫描电镜照片;
24.图4-b示出本发明对比例1制得产物的扫描电镜放大照片;
25.图5示出本发明实施例1-3和对比例1制得产物的xrd图谱。
具体实施方式
26.下面将对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。
27.本发明的第一方面在于提供一种纳米单晶三元材料,其由前驱体、锂源和含a元素的化合物混合烧结后与含m元素的化合物湿法包覆,最后经喷雾干燥制得。
28.所述前驱体选自含镍钴锰的氢氧化物、氧化物和碳酸盐中的一种或几种;优选地,所述前驱体选自含镍钴锰的氢氧化物和氧化物中的一种或两种;更优选地,所述前驱体为含镍钴锰的氢氧化物。
29.所述锂源选自含锂的碳酸盐、硫酸盐、硝酸盐、氧化物和氢氧化物中的一种或几
种,优选地,所述锂源选自含锂的碳酸盐、氧化物和氢氧化物中的一种或几种,更优选地,所述锂源为碳酸锂。
30.所述a元素选自mg、al、ti、zr、w、nb、y、nd和ta中的一种或几种;优选选自mg、al、ti、zr和w中的一种或几种;更优选为mg、al、ti和zr中的一种或几种。
31.含a元素的化合物选自含a元素的氢氧化物、氧化物、碳酸盐和硫酸盐中的一种或几种,优选地,所述含a元素的化合物选自含a元素的氧化物、氢氧化物和碳酸盐中的一种或几种更优选的,所述含a元素的化合物为含a元素的氧化物。
32.在本发明中,所述含a元素的化合物为掺杂化合物,a元素作为掺杂元素进入制得三元材料的晶体结构中,增大材料的层状结构中的层间距,增大晶胞参数c值,更易于锂离子嵌入和脱出,提升材料的充放电倍率性能;同时稳定材料的晶格,减小材料过渡金属层发生阳离子混排的频率及材料在高倍率充放电过程中受到的内部应力,减少材料在高倍率循环后发生的微裂纹,减小产生副反应的新的界面,从而提升材料的循环性能。
33.所述m元素选自si、al、ti、zr和w中的一种或几种,优选选自si、al、ti和w中的一种或几种;更优选为si、al和ti中的一种或几种。
34.含m元素的化合物选自含m元素的有机盐、碳酸盐、氧化物和氢氧化物中的一种或几种,优选选自含m元素的氧化物、碳酸盐、氢氧化物和钛酸正丁酯中的一种或几种,更优选为二氧化硅、氧化铝和钛酸正丁酯中的一种或几种。
35.含m元素的化合物为包覆材料,经试验发现,通过在纳米单晶材料表面进行包覆,制得的正极材料具有更好的物理储存性能,使其在后期制得电池的过程中对工艺的要求降低,不但降低了制备成本,还有效提高了其倍率性能和循环性能。
36.所述前驱体中金属元素之和、锂源中锂元素、含a元素的化合物中a元素和含m元素的化合物中m元素的摩尔比为1:(0.5~2):(0.0005~0.01):(0.0005~0.02),优选为1:(0.9~1.1):(0.001~0.005):(0.001~0.005),更优选为1:(0.98~1.10):(0.002~0.004):(0.002~0.004)。本发明人发现,当前驱体中金属元素之和、锂源中锂元素、含a元素的化合物中a元素和含m元素的化合物中m元素的摩尔比为1:(0.5~2):(0.0005~0.01):(0.0005~0.02)时,制得的产物具有最优的倍率性能和循环稳定性。
37.所述纳米单晶三元材料可由式nliani
x
coymn
1-x-y-zaz
o2·
m表示,其中,n>200,0.98≤a≤1.10,0.3≤x≤0.60,0.20≤y≤0.35,z<0.02。
38.所述纳米单晶三元材料的一次颗粒中值粒径为100~1000nm。
39.由本发明所述纳米单晶三元材料制得的全电池,在2.2v、100c、10s循环1周的内阻为7~10mω,循环100000周内阻为9~11mω,循环200000周内阻为11~14mω,循环500000周内阻为19~25mω。
40.在本发明中,如制备本发明所述的纳米单晶三元材料,其由包括以下步骤的方法制备:
41.步骤1、将称量好的锂源、预处理后的前驱体、含a元素的化合物湿法研磨、混合、干燥;
42.步骤2、将步骤1制得混合物高温烧结后粉碎;
43.步骤3、步骤2烧结后的材料与含m元素的化合物进行湿法包覆后喷雾干燥;
44.步骤4、将步骤3制得材料烧结。
45.步骤1中,前驱体的预处理温度为300~500℃,预处理时间为2~20h。
46.在步骤2中,所述高温烧结温度为700~1000℃,高温烧结时间为4~20h。
47.步骤3中,将湿法包覆后的材料进行喷雾干燥,喷雾温度为80~200℃。
48.步骤4中,烧结温度为300~600℃,烧结时间为1~10h。
49.本发明的第二方面在于提供一种根据本发明第一方面所述的纳米单晶三元材料的制备方法,所述方法包括以下步骤:
50.步骤1、将称量好的锂源、预处理后的前驱体、含a元素的化合物湿法研磨、混合、干燥;
51.步骤2、将步骤1制得混合物高温烧结后粉碎;
52.步骤3、步骤2烧结后的材料与含m元素的化合物进行湿法包覆后喷雾干燥;
53.步骤4、将步骤3制得材料烧结。
54.以下对该步骤进行具体描述和说明。
55.步骤1、将称量好的锂源、预处理后的前驱体、含a元素的化合物湿法研磨、混合、干燥。
56.在本发明步骤1中,所述前驱体选自含镍钴锰的氢氧化物、氧化物和碳酸盐中的一种或几种;优选地,所述前驱体选自含镍钴锰的氢氧化物和氧化物中的一种或两种;更优选地,所述前驱体为含镍钴锰的氢氧化物。
57.根据本发明,所述前驱体的预处理优选为低温预处理,该低温预处理优选在马弗炉中进行。预处理温度为300~500℃,优选为400~500℃,更优选为400~450℃。
58.所述预处理时间为2~20h,优选为5~15h,更优选为10~15h,例如12h。经试验发现,对前驱体进行低温预处理后制得的正极材料粒径较小,粒径大小更均一,其具有更良好的电化学性能,特别是循环稳定性显著提高。
59.所述锂源选自含锂的碳酸盐、硫酸盐、硝酸盐、氧化物和氢氧化物中的一种或几种,优选地,所述锂源选自含锂的碳酸盐、氧化物和氢氧化物中的一种或几种,更优选地,所述锂源为碳酸锂。
60.所述a元素选自mg、al、ti、zr、w、nb、y、nd和ta中的一种或几种;优选选自mg、al、ti、zr和w中的一种或几种;更优选为mg、al、ti和zr中的一种或几种。
61.含a元素的化合物选自含a元素的氢氧化物、氧化物、碳酸盐和硫酸盐中的一种或几种,优选地,所述含a元素的化合物选自含a元素的氧化物、氢氧化物和碳酸盐中的一种或几种更优选的,所述含a元素的化合物为含a元素的氧化物。
62.所述前驱体中金属元素之和、锂源中锂元素和含a元素化合物中a元素的摩尔比为1:(0.5~2):(0.0005~0.01),优选为1:(0.9~1.1):(0.001~0.005),更优选为1:(0.98~1.10):(0.002~0.004)。
63.将上述混合料溶于溶剂中制成浆料后,再置于砂磨机中进行湿法研磨,经试验发现,湿法研磨可有效提高最终制得正极材料的粒径均一性,材料的循环稳定性也有效提高,这可能是由于经湿法研磨后,含a元素的化合物与前驱体混合更均匀,阳离子混排显著降低,更有利于提高材料的倍率性能和循环稳定性。
64.所述溶剂选自水和乙醇中的一种或两种,优选为乙醇。所述研磨速度为2000~3000rpm,优选为2300~2800rmp,更优选为2500rmp。研磨时间为0.5~4h,优选为1~3h,更
优选为2h。若研磨速度太快,研磨时间太短,混合料混合后的均匀性和粒度均一性均较差,研磨速度太慢,研磨时间太长,则会降低制备效率。
65.混合研磨后的混合料进行干燥,所述干燥优选为真空干燥,干燥温度为80~150℃,干燥时间为5~15h,优选地,所述干燥温度为100℃,干燥时间为8h。真空干燥的目的主要为蒸发除去混合浆料中的溶剂,经试验发现,当干燥温度为80~120℃,干燥时间为5~15h时,兼具较优的干燥效果和较高的干燥效率。
66.最后将干燥后的物料研磨成粉末。所述研磨后的物料粒径为100nm~1000nm
67.步骤2、将步骤1制得混合物高温烧结后粉碎。
68.所述高温烧结在马弗炉中进行,高温烧结氛围为空气、氧气或空气与氧气任意比例的混合气,优选为空气或氧气气氛。
69.高温烧结温度为700~1000℃,烧结时间为4~20h;优选地,高温烧结温度为800~1000℃,烧结时间为5~15h;更优选地,高温烧结温度为850~950℃,烧结时间为8~12h。
70.烧结温度和烧结时间会影响最终制得三元材料的电化学性能,经试验发现,烧结温度高于1000℃,烧结时间超过20h,易生成缺氧型化合物,还会造成二次结晶,降低正极材料的比表面积,不利于锂离子的脱嵌,最终导致电容量、倍率性能下降,若烧结温度低于700℃,烧结时间短于4h,会造成反应不完全,晶体结构不能稳定生长,最终导致制得的材料晶体结构不完整,容易产生杂相,结晶度较低,使电池在充放电过程中的结构稳定性差,导致电化学性能降低。
71.将烧结后的产物进行粉碎,优选为机械粉碎,更优选为在气流粉碎机中进行粉碎。粉碎后的产物中值粒径为100~600nm。
72.步骤3、步骤2烧结后的材料与含m元素的化合物进行湿法包覆后喷雾干燥。
73.所述m元素选自si、al、ti、zr和w,优选选自si、al、ti和w中的一种或几种;更优选为si、al和ti中的一种或几种。
74.含m元素的化合物选自含m元素的有机盐、碳酸盐、氧化物和氢氧化物中的一种或几种,优选选自含m元素的氧化物、碳酸盐、氢氧化物和钛酸正丁酯中的一种或几种,更优选为二氧化硅、氧化铝和钛酸正丁酯中的一种或几种。
75.在本发明步骤3中,含m元素的化合物中m元素与锂源中锂元素的摩尔比为(0.0005~0.02):(0.5~2),优选为(0.001~0.005):(0.9~1.1),最优选为(0.002~0.004):(0.98~1.10)。
76.将含m元素的化合物置于溶剂中进行溶解,所述溶剂选自水和有机溶剂中的一种或几种,优选选自水和乙醇中的一种或两种,更优选为水或乙醇。用水或乙醇作为溶剂可避免在后期进行除溶剂的过程,简化了制备过程。
77.在本发明中,对溶剂的用量不做限定,只要能完全溶解含m元素的化合物即可。
78.根据本发明,将步骤2中粉碎后的产物置于含有m元素化合物的溶液中进行湿法包覆,优选为磁力搅拌,搅拌时间为1~4h,优选为2h。经试验发现,当所用搅拌方式为磁力搅拌时,搅拌时间为2h时,可使包覆物质在基体材料上包覆的更加均匀,最终制得三元材料的电化学性能最优。
79.将湿法包覆后的上述材料进行喷雾干燥,经喷雾干燥后制得的材料,为松散的软团聚,其在电池浆料匀浆的过程中可以被打开,团聚将打开形成新的单晶的分散分布,避免
了应力集中造成开裂的可能性,使制得材料的倍率性能和循环稳定性进一步提高。
80.所述喷雾温度为80~200℃,优选为100~200℃,最优选为100~150℃。喷雾温度太高或太低都会影响最终制得三元材料的电化学性能,这可能是由于喷雾温度会影响制得三元材料的粒径大小,若喷雾温度太低,材料颗粒在允许的时间内不会变的足够干燥,材料颗粒容易造成团聚,使得粒径变大,电化学性能降低,反之喷雾温度太高,会造成“过干”现象,同样不利于电化学性能的提高。
81.步骤4、将步骤3制得材料烧结。
82.将步骤3中喷雾干燥所得粉末进行二次烧结,烧结温度为300~600℃,烧结时间为1~10h;优选地,烧结温度为350~550℃,烧结时间为2~6h;更优选地,烧结温度为375~550℃,烧结时间为3~4h。
83.烧结温度和时间会影响最终制得正极材料的电化学性能,烧结温度高于600℃,烧结时间多于10h,正极材料表面的包覆材料可能会形成体相掺杂,使正极材料的晶体结构产生缺陷,导致由其制得电池的倍率性能和循环性能降低,同时还会延长制备时间,降低制备效率。若烧结温度太低,低于300℃,烧结时间太短,表面包覆材料的晶体结构生长不完全,电化学性能的下降。
84.将烧结后的产物进行机械粉碎,粉碎后的产物粒径为0.1~15μm,优选地,产物粒径为0.1~5μm,更优选地,产物粒径为0.1~1μm。
85.粒径过大或过小都会影响最终制得的三元材料的电化学性能,若粒径太大,在使用材料制备全电池的过程中制浆环节,材料不容易被分散成纳米颗粒;若粒径太小,正极材料的比表面积增加,则制备的材料在制成环节暴露的表面太多,与空气中的水和二氧化碳产生副反应,影响材料的后续使用,经试验发现,当产物粒径为3~10μm时,制得的三元材料具有较好的工程性能。
86.本发明所具有的有益效果:
87.(1)本发明所述的纳米单晶三元材料制备成本低,制备方法简单,具有优良的物理储存性能,对后期制成电池过程中工艺特别是湿度控制要求降低;
88.(2)本发明所述的纳米单晶三元材料采用喷雾的方法生成二次团聚体,避免其在循环过程中出现应力集中,减少开裂可能性,有效提高了其循环性能;
89.(3)本发明所述的纳米单晶三元材料,通过湿法均匀地在纳米颗粒表面形成包覆层,实现了锂离子的快速传导,倍率性能提高;
90.(4)本发明所述的纳米单晶三元材料在工艺上创新性的使用喷雾制备二次松散的团聚体,使材料具备较好的物理储存性能,在材料制备浆料涂布环节,团聚将打开形成新的单晶的分散分布,使材料具备更好的倍率性能和循环性能。
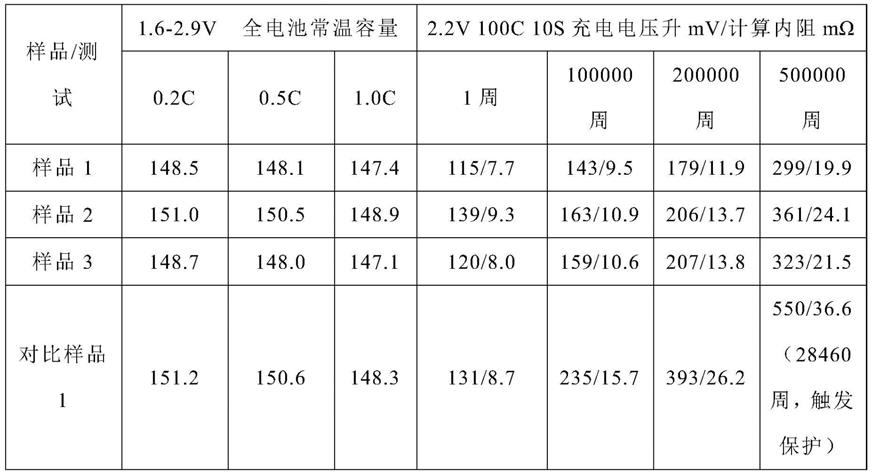
91.实施例
92.以下通过具体实例进一步阐述本发明,这些实施例仅限于说明本发明,而不用于限制本发明范围。
93.实施例1
94.将氢氧化物前驱体ni
0.333
co
0.333
mn
0.333
(oh)
2 150.00g,在400℃处理12h,加入碳酸锂64.89g,加入0.404g二氧化锆,加入100g无水乙醇,制备成浆料后在砂磨机上2500rpm研磨混合2h,在100℃下真空干燥8h,将干燥后物料研磨成粉末,放入刚玉坩埚中,在马弗炉
925℃处理10h,在气流粉碎机中粉碎,得到粉碎后物料。取二氧化硅含量20%的硅溶胶0.829g,加入80ml去离子水稀释,加入100.00g粉碎后物料,磁力搅拌2h,在120℃喷雾干燥,将喷雾干燥所得粉末在375℃高温处理4h,机械粉碎,得到样品1。样品1的化学组成式为:li
1.070
ni
0.333
co
0.333
mn
0.333
zr
0.002
o2·
0.0025sio2。
95.实施例2
96.将氢氧化物前驱体ni
0.500
co
0.250
mn
0.250
(oh)
2 150.00g,在420℃处理12h,加入碳酸锂63.47g,加入0.392g二氧化钛,加入100g无水乙醇,制备成浆料后在砂磨机上2500rpm研磨混合2h,在100℃下真空干燥8h,将干燥后物料研磨成粉末,放入刚玉坩埚中,在马弗炉920℃处理12h,在气流粉碎机中粉碎,得到粉碎后物料。取氧化铝含量20%的硅溶胶0.831g,加入80ml去离子水稀释,加入100.00g粉碎后物料,磁力搅拌2h,在150℃喷雾干燥,将喷雾干燥所得粉末在550℃高温处理4h,机械粉碎,得到样品2。样品2的化学组成式为:li
1.050
ni
0.500
co
0.250
mn
0.250
ti
0.003
o2·
0.0015al2o3。
97.实施例3
98.将氢氧化物前驱体ni
0.400
co
0.200
mn
0.400
(oh)
2 150.00g,在440℃处理12h,加入碳酸锂64.47g,加入0.1315g氧化镁,0.1677g氧化铝,加入100g无水乙醇,制备成浆料后在砂磨机上2500rpm研磨混合2h,在100℃下真空干燥8h,将干燥后物料研磨成粉末,放入刚玉坩埚中,在马弗炉930℃处理8h,在气流粉碎机中粉碎,得到粉碎后物料。取钛酸正丁酯1.115g,加入100ml无水乙醇,加入100.00g粉碎后物料,磁力搅拌2h,在100℃喷雾干燥,将喷雾干燥所得粉末在400℃高温处理4h,机械粉碎,得到样品3。样品3的化学组成式为:li
1.060
ni
0.400
co
0.200
mn
0.200
mg
0.002
al
0.002
o2·
0.003tio2。
99.对比例
100.对比例1
101.将氢氧化物前驱体ni
0.333
co
0.333
mn
0.333
(oh)
2 150.00g,加入碳酸锂64.89g,加入0.404g二氧化锆,加入100g无水乙醇,制备成浆料后,在100℃下真空干燥8h,将干燥后过300目筛,放入刚玉坩埚中,在马弗炉955℃处理10h,过300目筛,得到物料。取二氧化硅含量20%的硅溶胶0.829g,加入80ml去离子水稀释,加入100.00g粉碎后物料,磁力搅拌2h,在120℃喷雾干燥,将喷雾干燥所得粉末在375℃高温处理4h,机械粉碎,得到对比样品1。对比样品1的化学组成式为:li
1.070
ni
0.333
co
0.333
mn
0.333
zr
0.002
o2·
0.0025sio2。
102.实验例
103.实验例1全电池测试
104.软包电池制备:正极为20μm铝箔,单面100g/m2面密度双面涂布,92%材料,3%pvdf,2.5%ks,2.5%sp,60%固含量,nmp溶剂,极片压实密度3.2g/cm3,负极使用1c容量为165mah/g的li4ti5o
12
作为负极,使用20μm铝箔,单面110g/m2面密度双面涂布,90%材料,4%pvdf,6%sp,55%固含量,nmp溶剂,极片压实密度1.35g/cm3。电池叠片制备设计容量0.15ah的单片软包电池。按顺序在25℃完成各3周1.6-2.9v的0.2c循环、0.5c循环和1c循环后,将电池1c充电至2.2v,恒压30min,视为电池的起始状态。在此状态下,100c充电10s,静置30s,100c放电10s,静置30s,100c放电10s,静置30s,100c充电10s,静置30s,2.2v恒压60s,视为一个循环。模拟hev高倍率充放电工况。测试结果如表3所示。
105.表3全电池测试结果
[0106][0107]
从表3中可以看出,由本发明所述纳米单晶三元材料制得的全电池,在2.2v、100c、10s循环1周后其内阻为7~10mω,电压升高115~140mv;循环100000周后其内阻为9~11mω,电压升高140~165mv;循环200000周后内阻为11~14mω,电压升高175~210mv;循环500000周后其内阻为19~25mω,电压升高295~365mv,与对比例1相比,用本发明所述方法制得的纳米单晶三元材料的内阻增长和电压升高缓慢,循环稳定性提高。
[0108]
还可以看出,由本发明所述纳米单晶三元材料制得的全电池,在1.6~2.9v、25℃、0.2c的容量为148mah~151mah,0.5c的容量为148mah~151mah,1.0c的容量为147~149mah,倍率性能提高。
[0109]
实验例2xrd测试
[0110]
对实施例1-3和对比例1进行xrd测试,结果如图5和表1所示。
[0111]
由xrd衍射谱图可以看出,本发明制得的纳米单晶三元材料晶体生长较完全,且在图谱上未显示其它杂质相。
[0112]
表1xrd测试结果
[0113][0114]
从表1中可以看出,本发明所述制备方法制得产物晶粒尺寸增大,阳离子混排的频率显著降低。
[0115]
实验例3扫描电镜测试
[0116]
对实施例1-3和对比例1制得产物进行扫描电镜测试,实施例1制得产物的测试结
果如图1-a和图1-b所示,实施例2制得产物的测试结果如图2-a和图2-b所示,实施例3制得产物的测试结果如图3-a和图3-b所示,对比例1制得产物的测试结果如图4-a和图4-b所示。
[0117]
从图1-a、1-b、2-a、2-b、3-a和3-b中可以看出,实施例1,实施例2,实施例3制备的材料粒径大小均匀,粒径较小,大小范围在0.1~1μm之间,而从对比例1对应的图4-a和4-b中可以看出,对比例1中制得产物粒径较大,直径范围0.1~10μm之间,粒径大小不均,还可看出制备的材料为多晶二次颗粒材料。
[0118]
实验例4icp测试(单位为wt%)
[0119]
对实施例1、实施例2、实施例3和对比例1进行icp测试,测试结果如表2所示。
[0120]
表2icp测试结果
[0121]
样品linicomnzrtimgalsi实施例17.64920.1320.1718.760.18590.00230.00350.00290.0713实施例27.49330.1715.1914.160.00060.14650.00480.08310.0033实施例37.55924.1612.1722.490.00080.14880.05060.05550.0026对比例17.67020.1320.2618.850.18970.00160.00510.00350.0747
[0122]
考虑到icp测试的误差,测试结果可以说明材料的组成式分别为以上实施例1-3和对比例1中的组成式。
[0123]
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1