基于表面改性的碳的电化学电容器的电极的制作方法
基于表面改性的碳的电化学电容器的电极
1.早在1990年代就报道了借助重氮化学对碳材料进行改性,以在碳材料表面和各种官能团之间形成共价键。参见例如均以cabot corporation名义的us5554739和us5851280。
2.用于制备储能装置(例如电化学电容器和电池)电极的碳材料可以特别受益于通过重氮化学进行的表面改性,以使氧化还原分子掺入到活性炭和碳布等中,从而提高由这种形式的碳制成的电极性能。在us6,522,522(cabot corporation)中,关于重氮介导的碳表面改性工作已扩展到电极制造,并且在ep2886537、assresahegn等人[carbon 92(2015)362-381]、cougnon等人[journal of power source,274(2015)551-559]、comte等人[journal of materials chemistry a 3(2015),6146-6156]、us 2018/0182566、malka等人[在journal of the electrochemical society 165(14)a3342-a3349(2018)和journal of the electrochemical society 166(6)a1147-a1153(2019)中]报道了该领域的进一步努力。
[0003]
重氮盐的制备及其与碳材料的反应有不同的方法。该盐可以通过伯胺与亚硝酸在水性体系中(例如在nano2/hcl/h2o中)、或在有机溶剂中例如在有机亚硝酸盐如亚硝酸叔丁酯的帮助下反应形成。如us5,554,729的工作实施例中所示,盐可以预先在溶液中制备并且无需分离即可使用。重氮盐的分离和纯化也是可能的—参见ep2886537,其中以四氟硼酸盐的形式收集重氮盐,然后以电化学或化学方式将其接枝到碳上。
[0004]
一种似乎非常适合大规模生产电极的简洁方法由从伯胺前体原位合成重氮盐组成,其中盐形成反应在碳材料存在下发生,具有自发/化学还原和共价接枝到碳材料上。该方法通过以下方案示意性地举例说明:
[0005][0006]
(diazotation重氮化;spontaneous radical formation自发自由基形成;coupling to activated carbon by radical reduction通过自由基还原与活性炭偶联)
[0007]
合成途径的有效性在于其简易性:它既不涉及预先制备盐,也不涉及接枝诱导,例如没有重氮的电化学还原。
[0008]
胺向重氮的转化可以在nano2/hcl/h2o系统中或在有机介质中进行。
[0009]
例如,在us5,554,739中,使用等摩尔量的胺和nano2,并将冷的重氮溶液添加到碳材料的悬浮液中。
[0010]
在us6,522,522的实施例1中,通过将炭黑添加到2-氨基-蒽醌和盐酸的溶液中来制备电极材料。亚硝酸钠是最后加入的试剂,即以水溶液的形式逐渐以滴加的方式加入到反应混合物中。
[0011]
在journal of power sources(2015,同上)中,碳分散在乙腈中,并且伯胺(3,4-二甲氧基苯胺;相对于碳的0.3当量)与有机亚硝酸盐(亚硝酸叔丁酯,相对于碳的0.9当量)
一起加入。
[0012]
在journal of materials chemistry a(2015,同上)中,将伯胺(2-氨基-9,10-菲醌)溶解在乙腈中,然后加入1当量的亚硝酸叔丁酯、碳材料,然后再加入2当量的亚硝酸叔丁酯(两等份,超过30分钟)。
[0013]
在us2018/0182566的实施例1中,将有机亚硝酸盐(亚硝酸叔丁酯)溶解在乙腈中,并将溶液滴加到伯胺(4-氨基苯甲酸)和碳纤维的溶液中。4-氨基苯甲酸与亚硝酸盐的摩尔比为1∶3。
[0014]
在journal of the electrochemical society(2018,同上和2019,同上)中,将伯胺(分别为2-氨基蒽醌和3,4-二甲氧基苯胺)溶解在乙腈中,加入1当量的亚硝酸叔丁酯,然后加入碳材料,最后加入另一当量的亚硝酸叔丁酯。
[0015]
可以看出,尽管该方法很简单,但仍有不同的方法可以控制重氮盐的形成反应及其在碳质电极材料上的共价接枝。
[0016]
我们现在发现,通过仔细选择反应条件,即通过将碳材料与芳族伯胺和过量的亚硝酸盐的反应产物的溶液混合,可以通过重氮化学提高氧化还原分子在碳材料上的接枝度。即,使芳族伯胺与过量摩尔量的亚硝酸盐源在溶液中反应,随后将该溶液与碳材料混合。由于存在过量的亚硝酸盐,未反应的胺的量被最小化,结果碳表面的胺吸收被消除,使重氮的共价接枝可接近该表面。
[0017]
为支持本技术而进行的实验工作表明,所提出的工艺置换导致了有利的结果。如下所示,2-氨基蒽醌,研究重氮化学以提高电极性能的常用基准[另见us6,522,522和journal of the electrochemical society(2018,同上)],在有机溶剂中与活性炭结合。在下面报道的实验工作中,亚硝酸盐源以两种不同的方式提供给反应混合物:在添加碳材料之前,反应混合物中存在过量于2-氨基-蒽醌的亚硝酸盐;以及在添加碳材料之前,反应混合物中存在相对于2-氨基-蒽醌等摩尔量的亚硝酸盐;在添加完碳材料后,将剩余量的亚硝酸盐加入到反应混合物中。
[0018]
在下面报道的研究中,亚硝酸盐的总量是2-氨基蒽醌摩尔量的3倍。过量的亚硝酸盐量是1)在添加碳之前将其全部添加到2-氨基-蒽醌溶液中,或2)分成三等份,以30分钟的间隔依次加入溶液中,其中在第一部分亚硝酸盐之后添加碳。将反应物加入反应容器的顺序如下:
[0019]
1)2-氨基蒽醌
→
3eq.亚硝酸盐
→
碳材料
[0020]
2)2-氨基蒽醌
→
1eq.亚硝酸盐
→
碳材料
→
1eq.亚硝酸盐
→
1eq.亚硝酸盐
[0021]
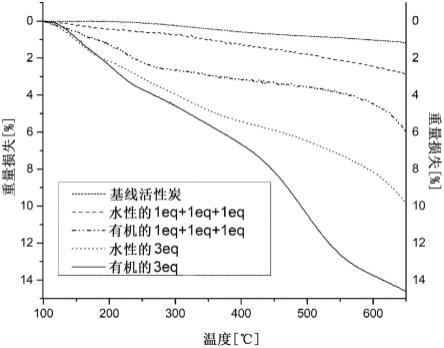
收集由表面改性的碳材料组成的产物,并通过热重分析(tga)进行测试,以确定质量相对于温度的变化,并通过上述方法测量接枝到碳表面上的醌衍生物的量。图1中显示的结果表明,在添加碳材料之前,当亚硝酸盐在反应混合物中过量存在时,接枝水平提高。独立于反应介质(水或有机物)观察到了相同的趋势。
[0022]
因此,本技术的一方面是一种制备表面改性的碳的方法,包括将碳材料加入到芳香伯胺与过量摩尔量亚硝酸源的反应产物的溶液中,并回收带有氧化还原活性位点的表面改性的碳。
[0023]
参与反应的芳香伯胺具有式(red)
n-ar-(nh2)m,其中:
[0024]
ar代表包含一个或多个环的芳族或共轭体系;
[0025]
m为1或2;
[0026]
red代表能够进行氧化还原反应的官能团,任选地为被保护的形式,其中red是连接到ar的一个或多个环,或者形成这些环的一部分;
[0027]
n是芳族伯胺中red基团的数量(1≤n≤5,例如,n=1、2或3)。
[0028]
表面改性的碳从反应混合物中回收,保护基团(如果存在)被去除,产物被加工并成型为电极,该电极可以组装以构建如下所述的储能装置。
[0029]
重氮盐的形成反应和接枝到碳上可以在水的酸性环境中进行,借助无机亚硝酸盐源,如亚硝酸碱金属盐,例如亚硝酸钠或亚硝酸钾,从而原位产生亚硝酸以驱动反应。酸性环境是由强无机酸(例如盐酸、硫酸和硝酸)产生的。优选的系统是nano2/hcl水溶液。反应在0至6(例如0至3)范围内的ph值和10至70℃范围内的温度(例如室温)下进行良好。为了完成接枝,能够从重氮离子中释放出氮(n2)并产生相应芳基自由基的还原剂也存在于反应混合物中。
[0030]
或者,盐形成反应和接枝到碳上发生在非水系统中,即在有机溶剂中。合适的有机溶剂是能够溶解胺和有机亚硝酸盐的惰性溶剂。可以使用极性非质子溶剂,如乙腈、丙酮、二甲基甲酰胺、二氧六环和二氧戊环。亚硝酸盐源为亚硝酸烷基酯,其中烷基为直链或支链c2-c7链,例如亚硝酸乙酯、亚硝酸叔丁酯和亚硝酸异戊酯。在有机体系中,重氮还原为相应的芳基自由基是自发发生的,即不需要施加电势或添加化学还原剂来强制重氮还原及其接枝。
[0031]
该方法的第一步是将芳族伯胺溶解在上述水性或有机介质中。芳族伯胺具有式(red)
n-ar-(nh2)m。芳核ar包含一个或多个芳环,包括杂芳环。当ar中存在两个或更多个环时,这些环可以稠合在一起形成多环芳族系统或可以桥接。因此,式(red)
n-ar-(nh2)m的芳族伯胺包括:
[0032]-被(red)n基团取代的苯胺;
[0033]-由稠合六元环组成的共轭多环,带有胺官能团并被(red)
x
基团取代,例如醌,例如衍生自萘(萘醌)、蒽(蒽醌)和菲(菲醌)的醌;
[0034]-由稠合的六元和五元杂芳环组成的芳族多环,例如含氮或含氧的芳环,例如稠合到咪唑环的吡啶或嘧啶环,带有胺官能团并被(red)n基团取代,或将(red)n作为环系统的一部分并入ar;
[0035]
氨基取代的联苯相关系统,具有连接苯环的连接基。
[0036]
现在转向(red)n官能团的定义,它最终解释了表面改性的电极碳材料中的氧化还原活性位点,red表示连接到环碳原子上的氧化还原基团,如:
[0037]
=o(氧代基团);
[0038]-oh(羟基),或被保护的羟基,如-op1,其中p1表示羟基保护基团,如烷基(例如甲基)、-si(r1)3,其中r1是烷基,尤其是甲基。即,羟基可以以相应的甲醚-och3、三甲基硅醚-o-si(ch3)3、或-och2och3(其中羟基被保护为甲氧基甲基醚)的形式被保护;
[0039]-sh(硫醇基)或被保护的硫醇,
[0040]-c(o)oh或其盐,例如其碱金属盐,或被保护的羧酸-c(o)op2,其中p2表示羧酸保护基团,例如,酸可以被保护为烷基酯,具体地为甲酯;
[0041]-so3h或其盐,例如其碱金属盐;
[0042]
应该注意的是,当两个或多个red基团存在于芳族伯胺中时,这些基团可以相同或不同。下面我们介绍几类优选的(red)
x-ar-(nh2)n起始材料。
[0043]
用于本技术的一类(red)
n-ar-(nh2)m化合物由(red)n取代的苯胺组成,其能够将氧化还原活性基团如硫醇、磺酸和羧酸合并到碳表面(在下面显示的方案中,碳表面由“梳状”结构表示):
[0044][0045]
上述(red)n取代的苯胺通过重氮途径接枝到碳表面上在有机溶剂中有效进行。例如,这些苯胺衍生物极易溶于乙腈。
[0046]
用于本技术的另一类优选的(red)
n-ar-(nh2)m化合物由(op1)n取代的苯胺组成(即ar是苯环,m=1,red是被保护的形式)。我们发现可以将一个或多个羟基接枝到碳表面,例如,在合适的保护基团的帮助下,每个芳环最多三个羟基。例如,通过将羟基作为相应的醚保护,例如甲基醚。下面说明了该方法,从(市售)3,4,5-三甲氧基苯胺开始。通过重氮途径接枝到碳表面发生在有机溶剂(例如乙腈)中,然后分离表面改性的碳并裂解保护基团以产生羟基氧化还原官能团,如下所示:
[0047][0048]
用于本技术的另一类优选的(red)
n-ar-(nh2)m化合物由(op1)
n1-(coop2)
n2-ar-(nh2)m组成,其中ar表示苯环,例如(op1)
n1-(coop2)
n2
取代的苯胺,其中op1是被保护的羟基,coop2是被保护的羧酸,n1和n2独立地为1或2。例如,市售的(oh)
n1-(cooh)
n2
取代的苯胺在有
机溶剂中被处理以将羟基作为甲硅烷基醚保护(例如,使用试剂(r1)3si-cl,例如其中r1是甲基,以将羟基作为三甲基甲硅烷基醚保护,使得p1优选为-si(ch3)3),以及将羧酸作为烷基酯保护(例如,使用合适的醇,例如甲醇,形成相应的甲酯,使得p2优选为-ch3)。所述保护反应可以在第一次保护反应后分离和纯化中间体依次进行。然而,一种更方便的方法由一锅法合成组成,其中起始材料(oh)
n1-(cooh)
n2
取代的苯胺与(r1)3si-cl和r1oh(r1独立为烷基)反应提供相应的保护的苯胺衍生物:
[0049]
(oh)
n1-(cooh)
n2
取代的苯胺
[0050]
(osi(r1)3)
n1-(coor1)
n2
取代的苯胺
[0051]
在加入过量的(r1)3si-cl和r1oh后,在有机溶剂如乙腈、无水四氢呋喃和二甲基甲酰胺中进行一锅法合成以形成被保护的苯胺衍生物。将被保护的苯胺衍生物从反应混合物中分离(例如,通过浓缩和过滤)并通过结晶纯化。然后纯化的(op1)
n1-(coop2)
n2
取代的苯胺进行根据本技术的重氮盐形成反应,例如通过溶解在有机溶剂中,与过量的亚硝酸盐反应并接枝到碳上。通过下述技术分离碳并裂解保护基团以恢复羟基和羧酸氧化还原活性官能团,最终产生所需的表面改性的碳产物,如下所示:
[0052][0053]
另一类有用的(red)
n-ar-(nh2)m化合物包括氨基取代的醌,其中red由并入醌体系中的酮基官能团提供(氧化还原活性是由于醌-氢醌对之间的相互转化)。这类(red)
n-ar-(nh2)m优选包括氨基取代的蒽醌(9,10-蒽醌和异构体)和氨基取代的菲醌(例如,2,7-二氨基菲-9,10-二酮)。后者的接枝如下图所示;其在乙腈等有机溶剂中表现良好。
[0054][0055]
另一类(red)
n-ar-(nh2)m由氨基取代的基于联苯的体系2hn-c6h
5-z-c6h
5-nh2组成,其中z表示结合氧化还原活性片段如二硫化物的连接基。一个实例如下所示:
[0056][0057]
氨基取代的杂环芳族化合物,例如由稠合的六元和五元杂环组成,具有~n=ch-nh~结构基序作为环体系的一部分,例如嘌呤:
[0058][0059]
(氨基可以连接在嘌呤的第2位或第6位),以及相应的氨基取代的杂环芳族化合物的共轭衍生物,其中稠合六元芳族环中的-ch=被-c(o)-官能团取代,例如嘌呤的6-酮衍生物,形成另一类有用的化合物,其可通过重氮途径接枝到碳上:
[0060][0061]
上述氨基取代的杂环芳族化合物在hcl水溶液中易于形成重氮盐。
[0062]
转向根据本技术的方法,将上述(red)
n-ar-(nh2)m起始材料(任选地以被保护的形式)溶解在酸性水介质或有机溶剂中。接着,将亚硝酸盐过量加入到芳族伯胺的水溶液/非水溶液中。最小过量为约1摩尔%(1.01摩尔当量的亚硝酸盐源),例如至少10摩尔%(1.10
摩尔当量的亚硝酸盐源),并且优选甚至显著更高的过量,高达50摩尔%(1.50摩尔当量的亚硝酸盐源),高达100摩尔%(2.0摩尔当量的亚硝酸盐源)。优选过量为100至200摩尔%,例如,加入2至3摩尔当量的亚硝酸盐源。
[0063]
如前所述,当反应在酸性水溶液中进行时,例如在盐酸中,还原剂有利于促进重氮离子还原为氮气(n2)和芳基自由基。因此可以将还原剂添加到水性反应混合物中(例如,由芳香伯胺在nano2/hcl/h2o中的溶液组成),例如铁、锌、铜和镍等金属,可以粉末等形式添加到溶液中。作为使用金属还原剂的替代方法,可以考虑使用还原剂如次磷酸(h3po2)和抗坏血酸。
[0064]
最后添加的试剂是要改性的碳材料。可通过本技术进行表面改性的合适碳材料包括活性炭、碳纤维、碳布和石墨粉。碳基质应主要包含sp2杂化碳,其具有尽可能大的表面积并且没有杂原子或金属污染物。例如,可以使用表面积为1500-2000m2/g的活性炭布。碳材料与(red)
n-ar-(nh2)m的质量比分别为10:0.1至0.1:10,例如1∶3-3∶1、2∶1-1:2、大约1∶1。
[0065]
在添加碳材料之后,将反应混合物保持一段时间,例如至少2小时。然后将表面改性的碳从反应混合物中分离,例如通过过滤,在有机溶剂(例如,dmf、甲醇和丙酮)中处理以去除过量的试剂(即,去除吸附的分子)并干燥。
[0066]
如上所述,在某些情况下,分子以电化学惰性形式接枝到碳上。即,在合成/接枝步骤中使用缺乏氧化还原活性的被保护的衍生物。因此,接枝后的分子需要转化为氧化还原活性化合物,例如,通过去除保护基团。一个典型的实例是氧化还原活性羟基取代的芳族化合物,其以被保护的形式负载到碳上,即作为醚化衍生物。即,(op1)
n-ar-nh2(如甲氧基取代的苯胺)用于接枝步骤,随后转化为氧化还原活性的(ho)
n-ar接枝的部分。
[0067]
该转化可以通过化学方式(例如,借助能够裂解保护基团的脱保护剂)或电化学方式实现。
[0068]
在从反应混合物中分离表面改性的碳之后,通过化学去除保护基团将接枝后的分子转化为其活性形式。表面改性的碳悬浮在惰性有机溶剂如二氯甲烷中,并借助合适的试剂完成保护基团的裂解。例如,可以借助三溴化硼(以二氯甲烷溶液出售)在二氯甲烷中实现脱烷基化,三溴化硼在室温下裂解甲氧基以形成羟基;使用氟化物源如nbu4nf(四丁基氟化铵;tbaf)或kf对甲硅烷基醚进行脱保护。
[0069]
在journal of power sources(2015,同上)中报道的条件下,通过电化学去除保护基团,将接枝后的分子转化为其氧化还原活性形式,例如将醚基转化为羟基。即,在由所述工作电极组成的三电极装置中,表面改性的碳被用作工作电极(并且如果需要,在粘合剂和增加导电的添加剂的帮助下成型),过量活性炭作为反电极,例如饱和甘汞电极(sce)作为参比电极。(op1)
n-ar接枝的部分(例如,p1是ch3)向氧化还原活性(ho)
n-ar接枝的部分的不可逆转化是在1-10m硫酸或1-10m hcl电解质溶液中、以1mv/s至10mv/s范围内的扫描速率在0-1v vs sce的δv范围内完成的。具有明确定义的质量平衡和电压窗口的双电极也可用于在商业规模上氧化大量物质。
[0070]
本文所述的具有共价结合到表面的氧化还原活性化合物的表面改性的碳可用作电化学电容器的电极材料。在浓盐酸、硫酸和氢氧化钾电解质溶液中使用三电极装置测量的循环伏安图(以2mv/s至20mv/s的各种速率记录),表明接枝分子产生的影响,改变了双电
层(edl)电容器表现,以并入表面上氧化还原分子的法拉第贡献;其中三电极装置中,表面改性的碳作为工作电极,未改性的碳作为反电极,饱和甘汞电极(sce)作为参比电极。记录显示表明存在明显的氧化还原位点的峰的伏安图,例如,对于:
[0071]
1)三羟基苯接枝的碳电极材料:
[0072][0073]
3)9,10-菲醌接枝的碳电极材料:
[0074][0075]
3)1,9-二氢-6h-嘌呤-6-酮接枝的碳电极材料:
[0076][0077]
4)2-羟基苯甲酸接枝的碳电极材料:
[0078][0079]
因此,本技术还涉及用本文所述的分子进行表面改性的碳材料(例如,活性炭、碳纤维、碳布和石墨粉),特别是上文刚刚提到的1)、2)、3)和4)变体,以及包含这种碳的电极。
[0080]
本技术还涉及具有上述分子的表面改性的碳,例如1)、2)、3)和4)变体,其中所述表面改性的碳的样品在氮气气氛下以每分钟10或20℃的升温速率,直到温度达到450℃进行的热重分析,表明重量损失至少15%,例如,至少20%;20%至30%、30%至40%、40%至50%、50%至60%、60%至70%、70%至80%、80%至90%、或90%至100%,表明接枝分子对碳材料的高负载。
[0081]
表面改性的碳,其中三羟基苯接枝到碳上,在tga中显示至少15%,例如至少20%,例如20%至50%,例如15%至30%的重量损失,直到温度达到450℃;以及
[0082]
表面改性的碳,其中9,10-菲醌接枝到碳上,在tga中显示至少20%,例如至少30%,例如30%至50%的重量损失(使用2,7-二氨基菲-9,10-二酮接枝到碳上);形成本技术的特定方面。
[0083]
基于本技术的表面改性的碳的电极通过本领域已知的技术制备。电极制备主要有两种方法:
[0084]
由活性炭纤维或布制成的单片电极,无需其他添加剂;即,单片电极被冲压并成型为所需的尺寸;以及
[0085]
复合电极,例如,当使用活性炭粉时,用改性的炭粉、粘合剂(ptfe在水中的悬浮液/pvdf等)和例如炭黑制备复合糊料以提高导电性。
[0086]
例如,为了制备复合电极,成分在以下浓度范围内组合:
[0087]
有效质量(本技术的改性的碳):70%-95%
[0088]
粘合剂(干重):4%-15%
[0089]
导电添加剂(炭黑、碳纳米管):0%-20%
[0090]
将所有成分与溶解粘合剂的溶剂,通常是醇(异丙醇或乙醇)或nmp(n-甲基吡咯烷酮),彻底混合形成均匀的浆液,然后将其部分干燥以形成糊状质地。然后将复合电极在滚压机中滚压至所需的电极宽度。
[0091]
本技术的电极可以组装成不对称电化学电容器,用作储能装置。因此,本技术还涉及一种电容器,其包括一对间隔开的电极、设置在所述电极和电解质溶液(例如hcl、h2so4或koh溶液)之间的空间中的隔板,其中所述电极中的一个包含本文所述的表面改性的碳。
[0092]
下面报告的实验结果表明本技术的一些电极材料,如9,10-菲醌接枝的碳电极(使用2,7-二氨基菲醌获得,如下所示):
[0093][0094]
在电化学电容器中表现良好,其电解质溶液含有盐而不是质子酸和氢氧化物碱,即由溶解在水中的盐制成,例如卤化物盐如溴化物和氯化物盐、例如碱金属卤化物(例如,溴化钠)或卤化铵、以及其他卤化物金属盐(例如二价卤化物,如氯化钡)、和硫酸盐例如碱金属硫酸盐。这种电解质溶液的浓度以重量计》10%,最高包括饱和极限。
[0095]
电容器通过本领域已知的方法组装,例如如上所述的具有单片/复合电极的对称或不对称电容器。电极放置在与集电器(例如,由碳片、金属箔或导电聚合物形成)或电池端子接触并用电解质溶液浸泡。放置在电容器中以将两个电极彼此分开的隔板的实例包括纤维素隔板(例如来自nkk)、塑料如高密度聚乙烯(hdpe)膜或多孔织物等。单个电池紧密闭合,以使每个电极与其集电器之间良好接触,并压在隔板上,以形成低电阻率的紧凑结构。单个电池堆叠在一起的双极配置可用于增加输出电压。例如,在us5,115,378、us5,581,438、us5,585,999和us6,522,522中描述了电化学电容器,显示了适用于本技术的整体双电层电容器设计。
实施例
[0096]
使用perkin elmer的tga-gc-ms(ei/ci)clarus 680/clarus sq 8c仪器进行热重分析(tga),以评估接枝部分的热稳定性。将6.5mg改性的碳布电极置于tga烘箱中,在氧化
铝坩埚中,在氮气气氛下以10或20℃/min的加热速率,从25至900℃(平衡吹扫80ml/min;样品吹扫20ml/min)。
[0097]
循环伏安法(cv)测量在biologic vsp稳压器中进行,并使用eclab软件进行分析。
[0098]
实施例1-2(本技术的)和3-4(对比)
[0099]
进行了一组实验以研究在添加碳之前,反应混合物中亚硝酸盐对2-氨基蒽醌过量存在的影响。
[0100]
实施例1的实验在有机体系中进行:
[0101]
将0.5gr 2-氨基蒽醌(2.2mmol)溶解在350ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯0.69gr(1ml 90%溶液)。5分钟后,加入1eq活性炭布。搅拌反应混合物24小时;然后过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0102]
实施例2的实验在水性体系中进行:
[0103]
将0.5gr 2-氨基蒽醌(2.2mmol)溶解在400ml 2m hcl中,直到胺衍生物完全溶解。加入3eq亚硝酸钠0.455gr。5分钟后加入1eq活性炭布。搅拌混合物24小时;然后过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0104]
实施例3的实验在有机体系中进行:
[0105]
将0.5gr 2-氨基蒽醌(2.2mmol)溶解在350ml acn中,直到胺衍生物完全溶解。加入1eq亚硝酸叔丁酯0.23gr(0.33ml 90%溶液),然后加入1eq活性炭。搅拌反应混合物30分钟,再加入1eq亚硝酸叔丁酯0.23gr(0.33ml 90%溶液),搅拌30分钟,然后再加入1eq亚硝酸叔丁酯0.23gr(0.33ml 90%溶液),搅拌24小时。过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0106]
实施例4的实验在水性体系中进行:
[0107]
将0.5gr 2-氨基蒽醌(2.2mmol)溶解在400ml 2m hcl中,直到胺衍生物完全溶解。添加1eq亚硝酸钠0.15gr,然后添加1eq活性炭。搅拌混合物30分钟,再加入1eq亚硝酸钠0.15gr,搅拌混合物30分钟,再加入1eq亚硝酸钠0.15gr,搅拌24小时。过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0108]
接下来,将每个样品放入热重分析仪中,显示质量随温度变化的热重曲线如图1所示(在氮气下,温度速率为20℃/min)。质量损失是由于接枝的醌衍生物从碳表面的消除和/或分解造成的,因此表明每个样品实现的接枝水平(即,重量损失百分比越大,接枝方法越好)。热重曲线证明了在添加碳之前存在于反应混合物中的过量亚硝酸盐对增加2-氨基蒽醌接枝到碳上的有利作用,实现了》14wt%的增加。
[0109]
实施例5a和5b
[0110]
使用3,4,5-三甲氧基苯胺对碳进行表面改性
[0111][0112]
a:将0.8gr 3,4,5-三甲氧基苯胺(4.4mmol)溶解在300ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯1.35gr(1.05ml 90%溶液)。5分钟后,加入1eq活性炭。搅拌混合物24小时。将活性炭布分成两部分并干燥。用150ml dcm悬浮1部分改性的活性炭。添加40ml在dcm中的1m bbr3溶液(3x3eq),并在室温下搅拌48小时。分批加入80ml甲醇,过滤并用水洗涤3次,和用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0113]
b:将3gr 3,4,5-三甲氧基苯胺(16mmol)溶解在600ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯5gr(7.1ml 90%溶液)。10分钟后,加入1.3g活性炭。在室温下搅拌混合物24小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时,得到1.93gr改性的碳(48%富集)。将活性炭布分成两部分并干燥。用150ml dcm悬浮1份改性的活性炭。添加40ml在dcm中的1m bbr3溶液(3x3eq),并在室温下搅拌48小时。分批加入80ml甲醇,过滤并用水洗涤3次,和用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0114]
实施例6
[0115]
使用4-氨基苯硫醇对碳进行表面改性
[0116][0117]
将1gr 4-氨基苯硫醇(8mmol)溶解在200ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯2.46gr(3.48ml 90%溶液)。5分钟后加入2eq活性炭布。在室温下搅拌混合物24小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0118]
实施例7
[0119]
使用4,4'-二硫基二苯胺对碳进行表面改性
[0120][0121]
将1gr 4,4'-二硫基二苯胺(4mmol)溶解在200ml acn中,直到胺衍生物完全溶解。添加3eq亚硝酸叔丁酯1.23gr(1.74ml 90%溶液)。5分钟后加入2eq活性炭。在室温下搅拌混合物24小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在
100℃下干燥4小时。
[0122]
实施例8
[0123]
使用5-氨基-2-羟基苯甲酸对碳进行表面改性
[0124][0125]
在250ml圆底烧瓶中,将3gr氨基水杨酸(19.5mmol)溶解在100ml无水thf中。缓慢加入2.34gr 2.75ml 1.1eq的tmsicl(21.45mmol)并搅拌1小时。加入60ml甲醇和2eq的tmsicl 4.95ml 4.2gr(39mmol)并搅拌24小时。浓缩反应混合物并溶解在dcm/etoac/己烷中。用水洗涤并从etoac/己烷中重结晶产物。
[0126]
将3.1gr保护的asa氨基水杨酸(13mmol)溶解在250ml acn中,直到asa完全溶解,加入3eq亚硝酸叔丁酯4.5gr(5.4ml 90%溶液),5分钟后加入1eq活性炭。搅拌混合物24小时。蒸发反应混合物并将其溶解在50ml dcm中并加入1eq bbr3,并在冰浴中搅拌3小时,在室温下搅拌24小时。用3eq水/甲醇淬灭bbr3。加入1eq tbaf并搅拌24小时。用水洗涤并过滤反应混合物,用等分量的乙腈、dmf、丙酮和甲醇洗涤。在100℃下干燥asa改性的碳4小时。
[0127]
实施例9
[0128]
使用5-氨基间苯二甲酸对碳进行表面改性
[0129][0130]
将1.6gr 5-氨基间苯二甲酸(8.8mmol)溶解在200ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯2.73gr(3.48ml 90%溶液)。5分钟后,加入2eq活性炭布。搅拌混合物24小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。在100℃下干燥改性的碳4小时。
[0131]
实施例10
[0132]
使用2,7-二氨基菲-9,10-二酮对碳进行表面改性
[0133][0134]
将3gr的2,7-二氨基菲-9,10-二酮(13.5mmol)溶解在350ml acn中,直到胺衍生物
完全溶解。加入3eq亚硝酸叔丁酯4.2gr(6ml 90%溶液)。10分钟后加入1.8gr活性炭布。将反应混合物搅拌24小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时,得到2.6gr改性的碳(40%富集)。
[0135]
实施例11
[0136]
使用5-氨基-2,3-二氢酞嗪-1,4-二酮对碳进行表面改性
[0137][0138]
将1.2gr 5-氨基-2,3-二氢酞嗪-1,4-二酮(6.9mmol)溶解在200ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯2.07gr(3ml 90%溶液)。5分钟后加入1eq活性炭布。将反应混合物搅拌24小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0139]
实施例12
[0140]
使用4-氨基苯磺酸对碳进行表面改性
[0141][0142]
将1gr 4-氨基苯磺酸(6.3mmol)溶解在200ml acn中,直到胺衍生物完全溶解。加入3eq亚硝酸叔丁酯1.95gr(2.76ml 90%溶液)。5分钟后加入2eq活性炭布,搅拌24小时。过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0143]
实施例13
[0144]
使用2-氨基-1,9-二氢-6h-嘌呤-6-酮对碳进行表面改性
[0145][0146]
将1gr的2-氨基-1,9-二氢-6h-嘌呤-6-酮(8mmol)溶解在150ml 1m hcl中,直到胺衍生物完全溶解。加入2eq亚硝酸钠(0.9gr)和fe粉(0.1gr),然后加入2eq活性炭布。在室温下搅拌混合物4小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0147]
实施例14
[0148]
使用7h-嘌呤-6-胺对碳进行表面改性
[0149][0150][0151]
将1gr 7h-嘌呤-6-胺(8mmol)溶解在150ml 1m hcl中,直到胺衍生物完全溶解。加入2eq亚硝酸钠(0.9gr)和铁粉(0.1gr),然后加入2eq活性炭。在室温下搅拌混合物4小时,过滤反应混合物并用等分量的乙腈、dmf、丙酮和甲醇洗涤。将改性的碳在100℃下干燥4小时。
[0152]
实施例15
[0153]
测试由表面改性的碳制成的电极
[0154]
使用循环伏安法(cv)评估电极的性能。cv在三电极装置中进行。工作电极是由改性的碳(直径4mm)制成的圆盘,平均重量为2mg(≈15mg/cm2)。反电极(直径11mm)由平均重量为11mg的未改性的碳制成。饱和甘汞电极(sce)用作参比电极。在真空下将电极浸入蒸馏水中,然后浸入电解质溶液中。通过将浸泡的电极引入玻璃碳集电器上组装三个电极t电池,并用隔板隔开。使用h2so4(1m、2m和4m)、hcl(1m、2m和4m)和koh(1m、2m和6m)作为电解质溶液。扫描速率在2mv/s到20mv/s的范围内。
[0155]
图2至11中所示的伏安图分别对应于实施例5至14,表明每个接枝分子将天然碳主体的正常性质从edl电容器性能改变为氧化还原分子在表面上的法拉第贡献(图2∶2m h2so4 3,4,5-三甲氧基苯胺;图3:4m koh 4-氨基苯硫醇;图4:4m koh 4,4'-二氨基二苯二硫;图5:6m h2so
4 5-氨基-2-羟基苯甲酸;图6:4m hcl 5-氨基间苯二甲酸;图7:6m koh 2,7-二氨基菲-9,10-二酮;图8:2m hcl 5-氨基-2,3-二氢酞嗪-1,4-二酮;图9:2m h2so
4 4-氨基苯磺酸;图10:1m hcl 2-氨基-1,9-二氢-6h-嘌呤-6-酮;图11:1m hcl-7h-嘌呤-6-胺)。
[0156]
实施例16
[0157]
使用3,4,5-三甲氧基苯胺将3,4,5-三羟基苯添加到碳布中,以及由此形成的电极的表征
[0158]
本实施例中报告的研究由两部分组成。在第一部分中,通过重氮基化学将3,4,5-三甲氧基苯胺(tma)添加到碳布中。该过程导致3,4,5-三甲氧基苯(tmb)接枝到碳上。然后分析tmb表面改性的碳布,以通过x射线光电子能谱(xps)确定表面组成,通过brunauer-emmett-teller(bet)方法确定比表面积,并通过热重分析(tga)确定样品随温度升高的重量损失。
[0159]
接下来,在第二部分中,对电化学惰性的3,4,5-三甲氧基苯进行脱保护以去除甲氧基,并得到相应的电活性3,4,5-三甲氧基苯(thb)。然后将thb表面改性的碳布作为电极材料进行测试。
[0160]
a部分:接枝和通过xps、bet和tga的表征
[0161][0162]
将100mg 3,4,5-三甲氧基苯胺(tma,0.546mmol)溶解在100ml乙腈中直至胺衍生物完全溶解。加入三当量的亚硝酸叔丁酯(0.240ml 90%溶液),然后加入100mg的kynol
tm
碳布。将混合物搅拌30分钟。将改性的布过滤并用等分量的二甲基甲酰胺、乙腈、丙酮和甲醇洗涤,并在60℃真空干燥,得到125mg改性的碳(添加25wt%的接枝分子,即3,4,5-三甲氧基苯)。
[0163]
通过x射线光电子能谱(xps)进行表面元素和化学状态分析。xps研究使用具有单色al kα源的thermo scientific nexsa光谱仪进行。测量光谱在200ev的pe和在50ev的高分辨率光谱下进行。使用了原位电荷中和,每组测量都相对在284.8ev的c1s进行了重新校准。由于样品的导电性质,在几十mev的范围内仅观察到轻微的带电。量化是在“智能”基线校正后使用nexsa的rsf进行的。使用gaussian-lorentian 70:30峰和合理的fwhm值进行峰拟合。
[0164]
光谱如图12a-f所示,排列如下:图12a和12d分别是未改性的和表面改性的碳布的c1s光谱;图12b和12e分别是未改性的和表面改性的碳布的o1s光谱;图12c和12f分别是未改性的和表面改性的碳布的n1s光谱。
[0165]
xps光谱证实了通过重氮化学将3,4,5-三甲氧基苯胺添加到碳布中:
[0166]
在图12d中看到的改性的碳的c1s光谱在286.5ev的结合能处表现出良好的分解和强峰,其被分配给附加的甲氧基(氧化碳),这是添加到碳上的接枝分子的一部分。在未改性的碳布的图12a中没有看到该峰。
[0167]
比较改性的(图12e)和未改性的碳布(图12b)的o1s光谱,可以看出o1s光谱中的接枝的碳布显示出比未改性的强得多的峰。这是由于每个接枝分子添加了三个氧原子。在533.5ev处,较高结合能峰的增强反映了较高电子密度氧位点的富集。这意味着氧在接枝电极中处于较高的氧化态。该结果表明,与来自接枝分子的甲氧基/羟基处的氧相比,源自碳表面的氧具有较低水平的结合能。
[0168]
正如预期的那样,在未改性的碳布中几乎没有检测到氮原子(图12c)。相比之下,tma接枝的碳布(图12f)在400ev附近表现出强氮峰。一个较小但仍可辨别的峰出现在稍低的结合能处,大约在402.2ev。这些峰可能是由于与重氮化学相关的交替接枝途径/副反应,在碳表面留下含氮部分,例如,重氮盐直接与碳底物偶联,而不是“正常”重氮化学,其导致自发降解为n2和芳基自由基,n2离开。然而,碳表面上氮的检测间接证明了接枝过程。
[0169]
bet测量在使用氮气作为吸附剂的quantachrome nova 3200e表面面积和孔径分析仪中进行。商业活性炭(kynol
tm
炭布)的表面面积约为1500m2/g。表1中列出的结果表明表面面积显著下降,证明了3,4,5-三甲氧基苯(tmb)的广泛接枝。
[0170]
表1
[0171][0172]
对于未改性的(商业碳布材料)和表面改性的碳布材料,使用上述仪器在氮气下以10k/min的升温速率进行tga。图13所附的热谱图表明tma表面改性的碳布样品在450℃的温度下重量损失约25%。即tga表明thb在活性炭上的质量负载量超过25%。
[0173]
b部分:电化学脱保护和表征
[0174]
接枝分子tmb经过电化学脱保护得到电活性tha分子。脱保护反应发生在三电极t电池中,以硫酸为电解质溶液,其中tma表面改性的碳布作为工作电极,随后进行电化学测量以评估tha接枝电极的性能。
[0175]
swagelok三电极t电池被用于电化学脱保护反应和后续测量。电极由ptfe圆柱体制成,该圆柱体与2、4毫米玻璃碳棒组装在一起,用作集电器和端子。工作电极由直径为6mm、平均重量为7mg(≈25mg/cm2)的改性的碳布冲压而成,其类似于商业设备中的真实电极,直径为2
×
12mm的反电极由平均重量为46mg(12mm双盘)的未改性的碳布冲压而成,导致电极质量负载约为40mg/cm2。nkk纤维素纸用作隔板。将电极浸入蒸馏水中并在真空下轻微旋转以实现良好润湿,然后浸泡在h2so4(2m)溶液中以使渗透压将溶液引入整个电极块内的孔中。饱和甘汞电极(sce)用作参比电极。
[0176]
不希望受理论束缚,甲氧基的电化学不可逆裂解(在2m硫酸电解质溶液中,在0.7v vs.sce),将非活性接枝tma分子转化为电活性tha,如以下方案所示:
[0177][0178]
(methyl sulfate:硫酸甲酯)
[0179]
极化显示在直到0.6v vs.sce的电位下被激活。氧的早期质子化引导贫硫酸根离子亲核试剂在带正电荷的甲基上发生e1反应。这个过程在图14a中接枝在碳布上的thb的循环伏安法中得到了很好的证明。tma接枝碳布的第一个循环伏安循环表明,在2mv/s的扫描速率下直到第三个循环,甲氧基裂解形成三羟基苯。在2m h2so4电解质溶液中以不同扫描速率记录的伏安图见图14b。与甲氧基裂解相关的峰值电流发生在0.6-0.7v,化合物在0.1-0.5v时转化为thb氧化还原活性。
[0180]
然后在设置为使用2m h2so4电解质溶液的三电极中通过恒电流测量研究thb改性的电极。图15a显示通过以3a/g的电流密度在-0.4v至0.8v的电压窗口上循环获得的充电/放电电压曲线。在2000次循环中测量的放电容量为65mah/g;然后它下降到55mah/g并在该值上保持额外的2500次循环。因此,总体而言,在kynol
tm
布上接枝的thb的恒电流测量和超过4500次循环的延长循环表明电极材料在酸性电解质中的良好性能。在0.1-0.5v vs.sce附近的稳定期表明法拉第氧化还原反应。
[0181]
恒电流测量的主要结果列于表2中,以及在electrochemical society 165(14)a3342-a3349(2018)中报告的蒽醌(aq)接枝的kynol
tm
布和在electrochemical society 166(6)a1147-a1153(2019)中报告的二羟基苯(dhb)接枝的kynol
tm
布的结果。结果表明,aq接枝的碳布、dhb接枝的碳布和thb接枝的碳布表现出相当的电容增加。
[0182]
表2
[0183][0184]
实施例17
[0185]
2,7-二氨基-9,10-菲醌添加到碳布中,以及由此形成的电极的表征
[0186]
本实施例中报告的研究由两部分组成。在第一部分中,通过重氮基化学将2,7-二氨基-9,10-菲醌(pq)添加到碳布中。然后对经pq表面改性的碳布进行分析,通过热重分析(tga)确定样品随温度升高的重量损失。
[0187]
接下来,在第二部分中,对pq表面改性的碳布作为电极材料进行了测试。
[0188]
a部分:接枝和通过tga的表征
[0189][0190]
重复实施例10中描述的流程。在上述条件下对样品(3-5mg)进行tga。热谱图附在图16中。还测试了由商用kynol
tm
布组成的参考样品。结果表明,直到~500℃,表面改性的碳的重量损失约为40-50%,这是由于添加到碳中的分子而增加的重量。
[0191]
b部分:电化学表征
[0192]
在中性电解质溶液(溴化钠和氯化钡)中,通过循环伏安法和恒电流测量,在前面实施例中描述的二/三电极电池中测试了pq接枝的碳布。
[0193]
图17a-c描述了在具有饱和溴化钠电解质溶液的两电极配置电池中进行的测试。
工作电极是市售的、未改性的布(直径为6mm;重量为5mg)。反电极是pq接枝的碳布(直径为12毫米;重量为35mg)。图17a中给出了以1mv和5mv的扫描速率产生的伏安图(分别为红线和蓝线)。对于恒电流测量,施加1a/g的电流密度。图17b中显示的结果表明了全电池的恒电流充放电电压曲线。延长循环(1000次循环)的性能如图17c所示,表明初始放电容量为35mah/g,能量密度为35wh/kg。
[0194]
图18a-c描述了在具有饱和氯化钡电解质溶液的三电极电池中进行的测试。工作电极是pq接枝的碳布(直径为6mm;重量为7mg)。反电极是市售的碳布(直径为12mm;重量为27mg)。以5mv的扫描速率产生的伏安图如图18a所示,表明了氧化还原反应的贡献。从图18b(以1a/g的电流密度获得),获得60mah/g的放电容量。在图18c中绘制了放电容量和效率与直到500次循环的循环次数的关系,表明在延长的循环中性能稳定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1