一种光响应型化疗药物共递送系统及其制备方法与流程
本发明涉及医药技术领域,特别涉及一种光响应型化疗药物共递送系统及其制备方法。
背景技术:
在癌症的化疗中,单一药物治疗往往达不到最佳的效果,基于多种药物同时给药的联合化疗(combinationchemotherapy)已经成为有效的解决方案之一。肿瘤的化疗失败很大一个原因就是肿瘤细胞产生了多药耐药(mdr)效应,而联合化疗是当前克服mdr的常用方法。联合化疗通常指2种或2种以上的药物同时给药,彼此产生协同作用得到1+1>2的治疗效果。与单一药物治疗相比,联合用药可以调节细胞不同的信号通路,在降低单一药物的用量及减轻副作用的同时有效地提高抗癌活性,克服肿瘤的耐药性。除了化学药物,肿瘤抗体及抗癌基因等也可以一起递送产生协同作用。目前,联合化疗已经成为癌症治疗的普遍方案。基于脂质体,高分子载体,纳米颗粒的共递送载药系统也已有文献报导。当然也各自有不少存在的问题,比如药物的突释现象;不同药物的顺序释放等。因此,设计出合理的药物共传递系统,在降低药物对正常组织的副作用同时有效抑制肿瘤细胞增殖,提升疗效,对当前癌症的治疗具有非常重要的理论意义及实用价值。
光敏载体可以利用光响应型化合物与化疗药物共轭结合生成前药,在体内受到光刺激后吸收光子产生化学反应,重新释放出活性药物,产生细胞毒作用。
此种治疗方法可利用外源激发光的非入侵性及极佳的操控性优点,对用药部位进行精准调控。是除光动力治疗外另一种有效的光学治疗方案。但是受到光敏载体活性位点的限制,普通的单个光敏载体分子只能携带一个药物分子,传递效率比较低,并无法直接应用于多药协同递送。
技术实现要素:
本发明的目的是提供一种光响应型化疗药物共递送系统及其制备方法。该光响应型化疗药物共递送系统利用光敏载体对化疗药物进行共价连接,制备成相应的前药,再利用光子优异的时空操控性,对给药部位直接激发,实现药物的定时、定点及顺序释放,协同作用而抑制肿瘤化疗中的mdr效应。
本发明的发明目的是由以下技术方案来实现的:
一种光响应型化疗药物共递送系统,它是含邻硝基苄基的双支化光敏载体同时搭载上不同作用机制的抗肿瘤药物,形成药物共递送系统,所述药物共递送系统的化学结构式为下述结构式ⅰ、ⅱ和ⅲ中的任意一种:
其中,r1和r2为两种不同的携带羧基的化疗药物;r为亲水的oeg(n)侧链,n为自然数,1≤n≤10;
其中,r3为携带羧基的化疗药物,r4为携带羟基或氨基的化疗药物;r为亲水的oeg(n)侧链,n为自然数,1≤n≤10;x为o或者n;
其中,r5和r6为两种不同的携带羟基的化疗药物,或者为两种不同的携带氨基的化疗药物;或者为一种携带羟基,另一种携带氨基的不同化疗药物;r为亲水的oeg(n)侧链,n为自然数,1≤n≤10;x1、x2均为o,或者x1、x2均为n,或者x1、x2中其中一个o,另一个为n。
进一步的,所述携带羧基的化疗药物为苯丁酸氮芥、美法仑、叶酸、甲氨蝶呤、阿里维a酸、倍萨罗丁和9-顺式视黄酸中的任意一种。
进一步的,所述携带羟基的化疗药物为姜黄素、去甲氧基姜黄素、双去甲氧基姜黄素、长春花碱、长春新碱、去甲长春花碱、喜树碱、10-羟基喜树碱、7-乙基喜树碱、康普瑞汀a4、依立替康、拓扑替康、吉西他滨、安西他滨、地西他宾、卡培他滨、曲沙他滨、紫杉醇、多烯紫杉醇、米托蒽醌和阿糖胞苷中的任意一种。
进一步的,所述携带氨基的化疗药物为阿霉素、表阿霉素、吡喃阿霉素、柔红霉素、氟脲嘧啶和喃氟啶中的任意一种。
上述的光响应型化疗药物共递送系统的制备方法,包括下述步骤:
一、制备邻硝基苄基为结构单元的双支化光敏载体:
(1)将4-位卤代的邻硝基苄醇通过钯催化偶联方法制成芳基硼酸酯;
(2)将含不同聚合度的乙二醇单甲醚先在甲苯中用对甲苯磺酰氯活化;将活化后的含寡聚乙二醇的对甲苯磺酸酯溶入二甲基亚砜中,在碳酸钾存在下与邻二卤代苯酚反应,得到亲水性的二卤代苯;
(3)将亲水性的二卤代苯与芳基硼酸酯用suzuki方法进行偶联,制备出亲水双支化的光敏载体;
二、制备两种化疗药物的共递送系统:
(1)当两种化疗药物均为携带羧基的化疗药物时,其制备方法如下:
a、将含邻硝基苄基的双支化光敏载体溶于有机相中,在缩合剂存在下,将第一种药物的羧基与邻硝基苄醇片段上的羟基进行脱水缩合,生成酯键;
b、将步骤a中的产物溶于有机相中,在缩合剂存在下,将第二种药物的羧基与另一个邻硝基苄醇片段上的羟基进行脱水缩合,得到目标产物;
(2)当两种化疗药物中一种为携带羧基的化疗药物,另一种为携带羟基或氨基的化疗药物时,其制备方法如下:
a、将含邻硝基苄基的双支化光敏载体通过苯环上的活性羟基与含羧基的化疗药物在缩合剂存在下脱水反应,制备出含一个药物分子的前药;
b、将步骤a中的产物与氯甲酸对硝基苯酯或二(对硝基苯基)碳酸酯反应,生成活性酯;
c、将步骤b中的产物活性酯与含羟基或氨基的化合物在碱催化下反应,得到目标产物;
(3)当两种化疗药物均为携带羟基的不同化疗药物,或者均为携带氨基的不同化疗药物;或者为一种携带羟基,另一种携带氨基的不同化疗药物时,其制备方法如下:
a、将含邻硝基苄基的双支化光敏载体与过量氯甲酸对硝基苯酯或二(对硝基苯基)碳酸酯反应生成双活性酯;
b、将步骤a中的产物双活性酯依次与两种药物在碱催化下反应,制备出目标产物。
进一步的,所述步骤一(1)中钯催化剂为四(三苯基膦)钯、醋酸钯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯和二(乙酰丙酮)钯中的任意一种。
进一步的,所述步骤一(2)中不同聚合度的乙二醇单甲醚为含2-11个乙二醇单体的寡聚乙二醇[oeg(n)],1≤n≤10。
进一步的,所述步骤二中的有机相为二氯甲烷、三氯甲烷、四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜和乙腈中的任意一种;缩合剂为二环己基碳二亚胺(dcc)、碳酰二咪唑(cdi)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edci)和2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)中的任意一种。
进一步的,所述步骤二中的碱为三乙胺、乙基二异丙基胺、n,n-二甲氨基吡啶、碳酸钾、碳酸钠、乙醇钠、叔丁醇钠和叔丁醇钾中的任意一种。
本发明的有益效果:
(1)本发明中的光响应型化疗药物共递送系统,是对邻硝基苄基类光敏载体进行了结构修饰,制备出双功能型载体,使之能够同时携带不同的化疗药物制备出抗肿瘤的前药,可以应用在肿瘤的治疗中。当前药被输送到病灶部位后,利用外源非破坏性的光子作为刺激因子,通过控制外源激发光的强度、时间、波长等参数,可以定时、定位地激发反应,释放出药物的活性分子;实现对药物的可控释放,使药物按照不同的作用机制协同工作,产生药物的协同治疗,提高肿瘤化疗的疗效及靶向性,具有较为广阔的应用价值。
(2)本发明中的光响应型化疗药物共递送系统,运用基于邻硝基苄基的双支化光敏载体,同时搭载上不同作用机制的抗肿瘤化疗药物,制备出一类光响应型抗肿瘤前药,建立了一种新型的化疗药物共递送系统,只运用一个载体分子,就可实现不同药物的同时传递。与高分子载体不同,本发明制备出的是小分子前药,并且具有亲水性的侧链,更有利于药物在水相生物体系中的分布。
(3)本发明中的光响应型化疗药物共递送系统由于采用的是光响应型载体,对ph和温度均不敏感,在生物体内还具有稳定性好,没有细胞毒性的优点。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为双支化光敏载体6(1)的合成路线图;
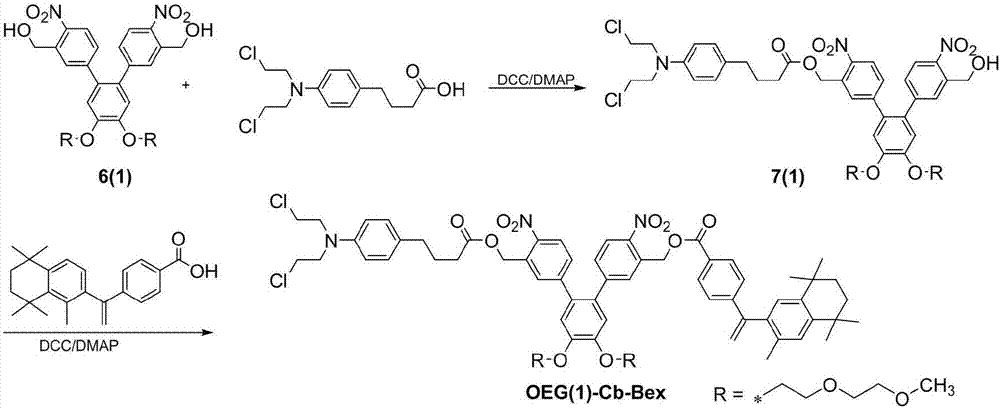
图2为实施例一中oeg(1)-cb-bex的制备路线图;
图3为实施例二中oeg(1)-cb-a4的制备路线图;
图4为实施例三中oeg(1)-dox-a4的制备路线图;
图5为实施例三中制备的oeg(1)-dox-a4的核磁共振氢谱图;
图6为实施例三中制备的oeg(1)-dox-a4的核磁共振碳谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
一、制备实施例:
1、双支化结构光敏载体的制备:
参考文献(tetrahedronletters,2016,57,959-963)所报导的方法,制备路线如图1所示,步骤如下:
(1)取20ml浓硝酸,在冰浴搅拌至0℃,移液管加入25ml浓硫酸,冰浴下搅拌20min,再加入10g间溴苯甲醛。室温下搅拌4h,加入少量水,用naoh调节ph为3-4。然后用乙酸乙酯萃取三次,有机相旋蒸浓缩得产物1;将产物1用25ml甲醇溶解,冰浴下分批加入2g硼氢化钠,室温下搅拌2h。在反应液中加入20ml水,然后用20ml乙酸乙酯萃取三次,有机相合并,无水硫酸钠干燥后旋蒸浓缩。粗产品过柱纯化,得11.29g产物2,产率90%;
(2)取0.5克产物2溶于5ml甲苯中,分批加入联硼酸频那醇酯0.66g,醋酸钾0.64g,室温搅拌5min后加入少量[1,1'-双(二苯基膦基)二茂铁]二氯化钯作催化剂,120℃回流4h。反应液加入10ml水,然后用10ml乙酸乙酯萃取三次,有机相合并,无水硫酸钠干燥后旋蒸浓缩。粗产物过柱纯化,得0.46g产物3,产率76%;
(3)取2.25g邻苯二酚溶于15ml三氯甲烷中,冰浴下分批加入2.6ml液溴,室温反应24h,冷却反应液。抽滤,并用三氯甲烷洗涤,得4.38g灰色固体产物4,产率80%;
(4)取二乙二醇单甲醚(r:n=1)先在甲苯中用对甲苯磺酰氯活化,将活化后的含寡聚乙二醇的对甲苯磺酸酯1.96g溶入二甲基亚砜中,加入0.87g产物4及2.70g碳酸钾,120℃反应7h。反应液加水10ml稀释,用稀盐酸调节ph为3-4,然后用乙酸乙酯萃取三次,有机相旋蒸浓缩后过柱纯化,得1.98g产物5(1),产率55%;
(5)取0.63g产物5(1)溶于甲苯,加入0.54g产物3,分别移取2mol/l的碳酸钠、无水乙醇1ml,加入[1,1'-双(二苯基膦基)二茂铁]二氯化钯作催化剂,120℃回流7h,反应液加水20ml,然后用乙酸乙酯萃取三次,有机相旋蒸浓缩后过柱,得0.2g双支化结构的光敏载体6(1),产率33%。
2、药物共递送系统的制备:
制备实施例一:
苯丁酸氮芥-蓓萨罗丁共递送系统[oeg(1)-cb-bex]的制备路线如图2所示,包括以下步骤:
(1)称取0.4g双支化结构的光敏载体6(1)溶于15ml二氯甲烷中,加入0.24g苯丁酸氮芥,0.2克二环已基碳二亚胺及少量的4-二甲胺基吡啶,室温搅拌反应4h;加入10ml水,用乙酸乙酯萃取三次,有机相浓缩后过柱纯化,得到0.29g黄色的产物7(1),产率50%;
(2)称取0.2克产物7(1)溶于15ml二氯甲烷中,加入0.1克蓓萨罗丁,70毫克二环已基碳二亚胺及少量的4-二甲胺基吡啶,室温搅拌反应4h;加入10ml水,用乙酸乙酯萃取三次,有机相浓缩后,残留物过柱纯化,得到0.22g黄色的产物oeg(1)-cb-bex,产率82%。
制备实施例二:
苯丁酸氮芥-康普瑞汀a4共递送系统[oeg(1)-cb-a4]的制备路线如图3所示,包括以下步骤:
(1)称取0.8g双支化结构的光敏载体6(1)溶于30ml二氯甲烷中,加入0.48g苯丁酸氮芥,0.4克二环已基碳二亚胺及少量的4-二甲胺基吡啶,室温搅拌反应4h;加入20ml水,用乙酸乙酯萃取三次,有机相浓缩后过柱纯化,得到0.58g黄色的产物7(1),产率50%;
(2)称取0.4克产物7(1)溶于15ml三氯甲烷中,加入氯甲酸对硝基苯酯0.13克,三乙胺0.2ml,室温下搅拌3h。反应液中加入20ml水,用15ml乙酸乙酯萃取三次,有机相合并后浓缩,残留物过柱纯化得0.38克产物8(1),产率80%;
(3)称取0.3克前一步产物8(1)溶于20ml三氯甲烷中,加入0.12克碳酸钾,0.1克康谱瑞汀a。室温下搅拌反应过夜。反应液浓缩后过柱纯化,得到0.3克目标产物oeg(1)-cb-a4,产率85%。
制备实施例三:
阿霉素-康普瑞汀a4共递送系统[oeg(1)-dox-a4]的制备,如图4所示,包括下述步骤:
(1)称取0.5g双支化结构的光敏载体6(1)溶于20ml三氯甲烷中,分批加入氯甲酸对硝基苯酯0.33克,完全溶解后,移液管加入三乙胺0.4ml,室温下搅拌3h,旋蒸上除去有机溶剂,残余黄色油状物柱层析纯化,得到0.69克黄色固体9(1),产率90%;
(2)称取0.6g步骤(1)得到的黄色固体9(1)溶于20ml二甲基亚砜中,加入0.2g康普瑞汀a4,边搅拌边加入碳酸钾0.18g,室温搅拌过夜;反应液加水20ml,用6m的盐酸调ph为3-4,然后用乙酸乙酯萃取三次,有机相减压浓缩后过柱纯化,得0.28克产物10(1),产率40%;
(3)称取0.2g步骤(2)得到的产物10(1)溶于无水n,n-二甲基甲酰胺中,加入盐酸阿霉素0.11g,加入三乙胺0.1ml,室温下搅拌过夜;反应液加20ml水稀释,乙酸乙酯萃取三次,有机相减压浓缩,残留物过柱纯化,得0.2g红黑色目标产物oeg(1)-dox-a4,产率74%。
本实施例制备得的中间体9(1)的1hnmr的谱图数据归纳如下,溶剂为氘代氯仿:1hnmr(400mhz,cdcl3)δ8.30(d,j=8hz,4h),8.05(d,j=8hz,2h),7.36-7.38(m,6h),7.29(dd,j=8hz,4hz,2h),7.04(s,2h),5.58(s,4h),4.27-4.29(m,4h),3.91-3.93(m,4h),3.73-3.76(m,4h),3.56-3.58(m,4h),3.37(s,6h)
中间体9(1)的13cnmr归纳如下,溶剂为氘代氯仿:13cnmr(101mhz,cdcl3)δ155.17,151.87,149.71,146.70,145.58,145.50,131.11,130.71,130.50,130.41,125.66,125.43,121.59,116.65,71.97,70.85,69.71,69.30,67.25,59.06
本实施例制备得的中间体10(1)的1hnmr的谱图数据归纳如下,溶剂为氘代氯仿:1hnmr(400mhz,cdcl3)δ8.28(d,j=8hz,2h),8.05(d,j=8hz,2h),7.45(dd,j=8hz,4hz,2h),7.33(d,j=8hz,2h),7.21-7.24(m,3h),7.13(d,j=8hz,1h),7.03-7.07(m,3h),6.83(d,j=8hz,1h),6.49(s,2h),6.45(d,j=8hz,1h),5.59(s,2h),5.54(s,2h),4.27-4.29(m,4h),3.90-3.93(m,4h),3.82(s,3h),3.74-3.76(m,4h),3.70(s,6h),3.66(s,3h),3.55-3.58(m,4h),3.37(s,6h)
中间体10(1)的13cnmr归纳如下,溶剂为氘代氯仿:13cnmr(101mhz,cdcl3)δ155.24,153.02,152.54,151.82,150.05,149.64,149.54,146.79,146.52,145.49,145.18,139.52,137.24,132.29,131.83,131.47,131.35,130.78,130.50,130.44,130.21,129.92,129.86,129.74,128.21,128.15,125.61,125.36,122.59,121.63,116.71,116.55,112.24,105.84,71.99,71.97,70.85,69.72,69.29,67.31,66.66,60.89,59.06,55.94,55.87
本实施例制备得的目标产物oeg(1)-dox-a4的1hnmr及13cnmr分别如图5、图6所示,溶剂为氘代氯仿。图5的谱图数据归纳如下:1hnmr(400mhz,cdcl3)δ13.98(s,1h),13.75(s,1h),8.02-8.04(m,2h),7.91(d,j=8hz,1h),7.78-7.80(m,1h),7.47(s,1h),7.38(d,j=8hz,1h),7.30(s,1h),7.01-7.14(m,6h),6.83(d,j=12hz,1h),6.45-6.49(m,4h),5.27-5.56(m,8h),4.76(s,2h),4.56(s,1h),4.27(s,4h),4.07-4.12(m,5h),3.57-3.91(m,27h),3.37(s,6h),2.96-3.05(m,2h),2.17(t,j=16hz,3h)
图6的谱图数据归纳如下:13cnmr(101mhz,cdcl3)δ214.06,187.15,186.76,161.13,156.29,155.73,154.94,153.07,152.76,150.12,149.54,146.80,145.65,145.29,139.64,137.26,135.88,135.55,133.68,132.42,131.81,131.50,130.34,130.13,130.01,128.37,128.24,125.63,125.39,125.25,122.75,120.92,119.94,118.56,116.97,116.65,112.34,111.66,111.48,105.94,72.09,72.03,70.92,70.85,69.81,69.37,67.44,66.88,65.67,63.33,61.01,59.16,59.13,56.78,56.04,52.14,47.26,36.04,35.78,34.06,32.04,31.55,30.43,29.82,29.44,29.36,27.33,25.64,22.81,16.96,14.25。
当然,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员应该可以根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明所附的权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!