疼痛缓解组合物的制作方法
本发明涉及一种疼痛缓解组合物。
背景技术:
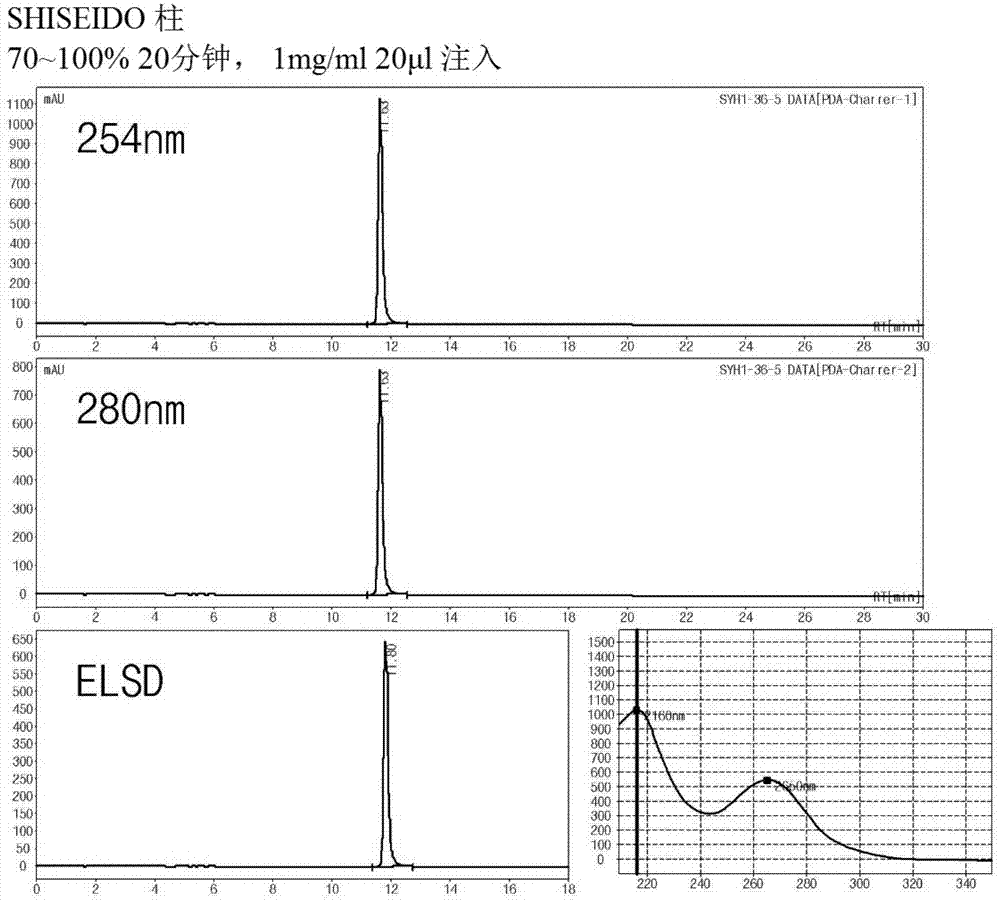
疼痛被定义为与实际或潜在的组织损伤相关的令人不快的感觉和情绪体验。在许多医学病症中,疼痛表现为主要症状。然而,由于疼痛是主观感知的,因此很难有效地诊断和治疗疼痛。疼痛导致患者严重的功能障碍,并危及其职业和社会功能以及家庭生活质量。可将疼痛大致归类为急性疼痛和慢性疼痛,例如神经性疼痛。急性疼痛通常由炎症或软组织损伤引起,并且负责炎症或软组织损伤的神经机制是众所周知的。因此,急性疼痛可以通过非甾体类抗炎药(nsaid)或一般镇痛药(如阿片类药物等)治疗。神经性疼痛通常由外周感觉神经损伤引起,并且神经性疼痛的强度非常严重。另外,由于神经性疼痛甚至在使用镇痛药(包括阿片类药物)后也经常复发,因此很难实现神经性疼痛的有效治疗。目前使用的镇痛药大致分为两类。第一类包括一组nsaid和与其相关的环加氧酶同种型2(cox-2)抑制剂,第二类包括鸦片制剂,例如吗啡。这些镇痛药对一般疼痛有效,但对某些特定类型的疼痛(如神经性疼痛)几乎没有镇痛作用。高剂量施用阿片类药物可能多少有助于缓解疼痛。然而,增加阿片类药物的剂量会引起严重的副作用或潜在的成瘾。由于nsaid具有比阿片类药物低的镇痛作用,因此应当施用增加的剂量以实现令人满意的镇痛作用,而且可以产生nsaid相关的胃肠道副作用。因此,迫切需要开发一种新的疼痛缓解化合物或类似物,其使用与目前已知的镇痛药机制不同的机制,可用于缓解疼痛而没有副作用。技术实现要素:技术问题本发明的实施方式提供了一种药物组合物,其可以快速抑制疼痛而没有副作用。技术方案根据本发明的一个方面,提供了一种疼痛缓解组合物,其包含作为有效成分的式1的松香-6,8,11,13-四烯-18-酸(abieta-6,8,11,13-tetraen-18-oicacid),或其药学上可接受的盐或溶剂化物:<式1>根据本发明的药物组合物使用从天然存在的植物-松果的碳化物提取物中分离的天然衍生的化合物,从而表现出优异的疼痛缓解效果而没有副作用。附图说明图1显示了根据实施例1从松果碳化物提取物中分离的式1化合物的高效液相色谱(hplc)色谱图。图2显示了根据实施例1从松果碳化物提取物中分离的式1化合物的lc/ms图谱。图3显示了根据实施例1从松果碳化物提取物中分离的式1化合物的1h-nmr图谱和13c-nmr图谱。图4是显示根据实施例2制备的松果碳化物提取物的组分的比较结果的图。图5是显示根据使用的各种提取溶剂,根据实施例2制备的松果碳化物提取物的组分的比较结果的图。图6是显示根据实施例2,根据样品与溶剂比(w/v)的松果碳化物提取物的组分的比较结果的图。图7是显示根据实施例2,根据提取时间的松果碳化物提取物的组分的比较结果的图。图8是显示trpv激动剂治疗对细胞内钙水平增加的影响以评估式1化合物的疼痛缓解功效的图。图9是根据提取溶剂比较提取量的图。图10是根据提取时间比较meoh提取量和水提取量的图。具体实施方式以下将更全面地描述本发明。然而,本发明可以以许多不同的形式实施,并且不应该被解释为限于本文所阐述的实施方式。本发明提供一种疼痛缓解组合物,其包含作为有效成分的式1的松香-6,8,11,13-四烯-18-酸,或其药学上可接受的盐或溶剂化物:<式1>在式1中,h可以全部被具有1-10个碳原子的烷基取代,并且所有这些取代基都意欲包括在本发明的范围内。根据本发明的组合物可以抑制各种类型的疼痛,特别是灼痛或外伤疼痛。因此,根据本发明的组合物可以抑制疼痛,特别是由皮肤损伤引起的疼痛。根据本发明的式1化合物及其药学上可接受的盐或溶剂化物可用于缓解疼痛,但根据本发明的式1化合物的用途不限于特定类型和强度(严重程度)的疼痛。根据本发明的式1化合物可以从松果碳化物提取物中分离。此外,所述药物组合物优选包含含有式1化合物的松果碳化物提取物。当以从松果碳化物提取物中分离的化合物或直接为包含式1化合物的松果碳化物提取物的形式使用时,式1化合物可以表现出优异的疼痛缓解效果而没有副作用。然而,式1化合物的制备方法不限于本文公开的方法。根据本发明的一个实施方式,松果碳化物提取物可以通过将水、具有1至4个碳原子的低级醇、丙酮、乙酸乙酯、氯仿或其混合溶剂加入到松果碳化物中来制备。优选地,通过将水、甲醇、乙醇或其混合溶剂加入到松果碳化物中来制备所述松果碳化物提取物。最优选地,通过添加100%甲醇作为提取溶剂来制备所述松果碳化物提取物,这提高了根据本发明的化合物中活性组分的含量。根据本发明的一个实施方式,优选地,通过以1(w)∶10-150(v)的比例将提取溶剂(v)添加到松果碳化物材料(w)中来制备所述松果碳化物提取物。如果提取溶剂的用量超过上述比例,则活性组分不能被充分提取。根据本发明的一个实施方式,所述松果碳化物提取物优选通过超声提取或回流提取获得。当使用水作为提取溶剂时,优选使用回流提取,而当使用有机溶剂作为提取溶剂时,优选使用超声提取。提取时间可以在10分钟至100分钟,优选20分钟至40分钟的范围内。即使提取时间延长,提取的活性成分的量也不会增加,如果提取时间太短,则不能提取足够量的活性成分。根据本发明的化合物可以以药学上可接受的盐的形式给药。本文所用的短语“药学上可接受的盐”是指由无毒或低毒的酸或碱制备的盐。当根据本发明的化合物含有相对酸性官能团时,通过使中性形式的化合物与足量的所需碱和适当的惰性溶剂接触,可以得到碱加成盐。药学上可接受的碱加成盐的实例包括但不限于锂盐、钠盐、钾盐、钙盐、铵盐、镁盐、有机氨基盐等。本发明的化合物可包括其溶剂化物,特别是水合物。此外,本发明的化合物可包括非溶剂化形式以及溶剂化形式(例如水合物)。本发明的化合物可以以结晶或无定形形式存在,并且这些物理形式包括在本发明的范围内。本发明提供一种药物组合物,其包含所述化合物或其药学上可接受的盐或溶剂化物,和药学上可接受的赋形剂或添加剂。根据本发明的化合物或其药学上可接受的盐/溶剂化物可以单独给药或与适当的载体或赋形剂组合给药。给药方式可以是单剂量或重复剂量。根据本发明的药物组合物可以是固体制剂或液体制剂。所述固体制剂可以采用粉末、颗粒、锭剂、胶囊、栓剂等形式,但不限于此。所述固体制剂可包含赋形剂、调味剂、粘合剂、防腐剂、崩解剂、润滑剂、填充剂等,但不限于此。所述液体制剂可以采用溶液(例如水或丙二醇溶液)、悬浮液、乳液等的形式,但不限于此。所述液体制剂可以通过添加适当的着色剂、调味剂、稳定剂、增粘剂等来制备。根据待治疗的疾病和受试者的病情,本发明的药物组合物可以以适于口服给药、注射(例如,肌内注射、腹膜内注射、静脉注射、输注、皮下注射或植入)、吸入、鼻内给药、阴道给药、直肠给药、舌下给药、透皮给药或局部给药的制剂形式给药,但不限于此。根据给药途径,本发明的药物组合物可以配制成通常使用且无毒的适当给药单位制剂,并且包括药学上可接受的载体、添加剂和媒介物。能够连续释放药物一段预定时间的贮库制剂也可包括在本发明的范围内。本发明还提供一种用于缓解疼痛的方法,包括向需要缓解疼痛的受试者施用治疗有效量的式1的化合物或其药学上可接受的盐或溶剂化物。在缓解疼痛的方法中,式1的化合物或其药学上可接受的盐或溶剂化物可以每天约0.1mg/kg至约1000mg/kg,优选约2.5mg/kg至约500mg/kg的量施用。然而,剂量可根据患者的状况(年龄、性别、体重等),所治疗病情的严重程度,所用化合物等而变化。如果需要,可以将化合物的有效日剂量分开并在一天中以适当的间隔多次给药。现将通过以下实施例进一步描述本发明,列出这些实施例以提供本发明的完整公开,但不意图限制本发明的范围。实施例1:松果碳化物提取物的制备和式1化合物的分离在室温下,将从韩国graeme有限公司收集的2.53kg松果碳化物用3.5lmeoh重复提取四次,然后浓缩,得到约92gmeoh提取物。分别使用正己烷和etoac对所得meoh提取物进行三次溶剂分馏,得到约6g正己烷提取物和约45getoac提取物。使用sephadexlh-20凝胶对etoac提取物进行柱色谱(chcl3-meoh,1∶1),得到5个馏分f1-f5。使用硅胶柱(正己烷-etoac,1∶0-0∶1)分离馏分f4,得到f4.1-f4.6。此后,使用硅胶柱(chcl3-meoh,100∶0-98∶2)用mplc(biorageisoleraiso-1sv)分离馏分f4.3,得到f4.3.1-f4.3.4。使用带有ymc-packoda-a柱(meoh-h2o,7∶3-1∶0,8ml/min)的制备型hplc(varianprostar210)纯化馏分f4.3.3以分离式1化合物。将分离的化合物进行hplc(varian920-lc)和uplc(watersacquityuplctm)以鉴定其纯度,并使用lc/ms(watersuplc/q-tofms系统)和gc/ms(agilent7890/5973ms系统)鉴定化合物的分子量。然后,使用nmr(核磁共振,varian系统500mhz)进行1h-、13c-、1h-1hcosy、hsqc和hmbcnmr的测量,并鉴定分离的化合物的结构,从而获得式1化合物:<式1>c20h26o2的分子量:298.42esi-msm/z:297[m-h]-.1h-nmr(500mhz,cdcl3):δ1.09(3h,s,h-20),1.23(3h,d,j=2hz,h-16),1.24(3h,d,j=2hz,h-17)1.42(3h,s,h-19),1.68-1.87(5h,m,h-1a,2,3),2.21(1h,d,j=13.5hz,h-1b),2.86(1h,m,h-15),2.92(1h,s,h-5)5.80(1h,d,j=10.5hz,h-6),6.53(1h,d,j=10.5hz,h-14),6.92(1h,d,j=2hz,h-9),7.08(2h,m,h-11,12).13c-nmr(125mhz,cdcl3):δ17.2(c-19),18.4(c-2),20.9(c-20),24.0(c-16),24.0(c-17),33.7(c-15),35.4(c-1),35.8(c-3),37.2(c-10),46.2(c-4),46.6(c-5),121.7(c-11),124.8(c-9),125.8(c-12),128.5(c-14),129.7(c-6),132.6(c-8),145.1(c-9),145.4(c-13),184.0(c-18).实施例2:根据提取条件对松果碳化物提取物进行比较在改变包括提取方法、提取溶剂类型、所用提取溶剂的组成和量以及提取时间在内的5种条件下,各取3g实施例1中使用的松果碳化物进行提取,在4000rpm下进行离心分离,过滤上清液,然后浓缩上清液以比较提取量,并使用uplc进行组分分析。采用回流提取和超声提取作为提取方法,使用水、乙醇和meoh溶剂作为提取溶剂。作为提取溶剂的meoh、etoh和水的组成在30%至100%的范围内变化,并且根据所用提取溶剂的类型,提取时间在10分钟至120分钟的范围内变化。在与实施例1相同的条件下进行分析,并在uv254nm鉴定每种提取物的纯度。提取方法:回流提取/超声提取使用的提取溶剂:甲醇、乙醇和水提取溶剂组成:30%、50%、70%和100%提取溶剂的量:30ml和100ml提取时间:10分钟、30分钟、60分钟、90分钟和120分钟-提取方法的比较在相同的提取条件下,使用meoh和水各100ml作为提取溶剂,通过超声提取和回流提取进行提取30分钟。当使用meoh作为提取溶剂时,发现根据提取方法的提取量没有显著差异。当使用水作为提取溶剂时,回流提取的提取量大于超声提取的提取量。在uplc分析中,没有发现在甲醇和水提取溶剂中根据提取方法存在特别的组分差异(参见图4)。[表1]-提取溶剂组成的比较为了分析根据提取溶剂组成的提取量的差异,使用meoh、etoh和水作为提取溶剂进行提取,同时将每种组成改变为30%、50%、70%和100%。在相同条件下,各自使用提取溶剂30ml通过超声提取进行提取30分钟。当使用meoh作为提取溶剂时,提取量大于在相同条件下使用etoh时的提取量。特别是,当使用100%meoh时,提取效率最高。在uplc分析中,在meoh和etoh之间,没有发现提取溶剂组成的实质差异。根据提取溶剂组成,极性和非极性组分之间的含量存在差异(参见图5和图9)。[表2]另外,通过使用meoh作为提取溶剂进行提取,比较根据样品与溶剂比(w/v)的提取量的差异。在相同条件下,使用1∶10(30ml)和1∶30(100ml)的样品-溶剂比(w/v)进行提取。提取结果证实,使用100mlmeoh时的提取量约为使用30mlmeoh时提取量的两倍或更多。没有发现组分差异(参见图6和图10)。[表3]提取量的比较-提取时间的比较为了比较根据提取时间的提取量,使用两种提取方法和对应于所述提取方法的合适提取溶剂进行实验。通过在10分钟、30分钟、60分钟和90分钟的四个提取时间条件下,使用30mlmeoh溶剂进行超声提取来比较提取量。通过在30分钟、60分钟、90分钟和120分钟的四个提取时间条件下,使用显示出相对高的提取效率的使用100mlh2o溶剂的回流提取来比较提取量和溶剂组分。除提取时间外,所有提取条件对于两种提取方法都保持相同。提取结果表明,提取量随提取时间的延长略有增加。仅发现提取量的微小差异,并且该差异被认为是统计学上不显著的。另外,发现根据提取时间的提取溶剂组成没有特别差异(参见图7和图10)。[表4]提取量的比较如上所述,可以理解,当使用水和低级醇时,可以以最高效率提取松果碳化物的活性组分。实验实施例1:式1化合物的活性测定为了研究实施例1中分离的式1化合物的活性,评估疼痛缓解效果。通过鉴定细胞内钙水平和信号通路证实了疼痛缓解。在对用式1化合物孵育24小时至50%汇合的白色大鼠中的f11感受伤害的感觉细胞系进行预处理后,测量trpv离子通道活性。用2-apb(2-氨基乙氧基二苯基硼酸酯)作为trpv离子通道的预活化剂处理f11细胞系,并且使用fluo-4-am荧光染料实时评估响应于2-apb处理而触发的细胞内钙变化。证实了,当用2-apb作为trpv离子通道的预活化剂处理f11细胞系时,在20秒内瞬时显示细胞内钙水平急剧增加,并且在大约5分钟内增加的细胞内钙水平恢复到基准水平。然而,当用式1化合物预处理f11细胞系24小时时,由2-apb诱导的细胞内钙水平的增加被强烈抑制,表明式1的化合物提供了疼痛缓解功效,如图8中以及以下制成表的式1化合物处理的实验组和未处理的对照组之间的比较数据所证实的那样(参见图8)。对照式11.24771.10311.25161.25031.68581.41311.58991.30081.62391.22511.5261.15362.023s1.1252.04041.15721.28951.12931.33811.17141.27421.16561.31741.4341.31671.86631.36361.38931.68122.18211.36781.75111.22661.56811.46211.73681.55621.29471.2439当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1